| ☐ |
REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934
|
| ☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
| ☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
| ☐ |
SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
Title of each class
|
Trading Symbol(s)
|
Name of each exchange on which registered
|
|
American Depositary Shares, each representing one ordinary share, nominal value £0.002 per share
|
IMCR
|
The Nasdaq Stock Market LLC
|
|
Ordinary share, nominal value £0.002 per share*
|
*
|
The Nasdaq Stock Market LLC*
|
|
Large accelerated filer ☐
|
Accelerated filer ☐
|
Non-accelerated filer ☒
|
Emerging growth company ☒
|
|
†
|
The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
|
|
Page
|
|||
|
PART I
|
|||
|
|
|||
|
ITEM 1.
|
5
|
||
|
ITEM 2.
|
5
|
||
|
ITEM 3.
|
5
|
||
|
A.
|
5
|
||
|
B.
|
5
|
||
|
C.
|
5
|
||
|
D.
|
5
|
||
|
ITEM 4.
|
89
|
||
|
A.
|
89
|
||
|
B.
|
90
|
||
|
C.
|
148
|
||
|
D.
|
148
|
||
|
ITEM 4A.
|
148
|
||
|
ITEM 5.
|
149
|
||
|
A.
|
149
|
||
|
B.
|
161
|
||
|
ITEM 6.
|
171
|
||
|
A.
|
171
|
||
|
B.
|
190
|
||
|
ITEM 7.
|
190
|
||
|
A.
|
190
|
||
|
ITEM 9.
|
196 | ||
|
A.
|
196 |
||
|
B.
|
196 |
||
|
C.
|
196 |
||
|
D.
|
196 |
||
|
E.
|
196
|
||
|
F.
|
196
|
||
|
ITEM 10.
|
196
|
||
|
|
A. |
196
|
|
|
|
B. |
196
|
|
|
|
C. |
196
|
|
|
|
D. |
197
|
|
|
|
E. |
197
|
|
|
|
F. |
205
|
|
|
|
G. |
205
|
|
|
|
H. |
205
|
|
|
|
I. |
206
|
|
|
|
|
|
|
|
ITEM 11.
|
206
|
||
|
ITEM 12.
|
207
|
||
|
|
A. |
207
|
|
|
|
B. |
207
|
|
|
|
C. |
207
|
|
|
|
D. |
207
|
|
|
|
209
|
||
|
ITEM 13.
|
209
|
||
|
ITEM 14.
|
209
|
||
|
ITEM 15.
|
209
|
||
|
|
A. |
209
|
|
|
|
B. |
210
|
|
|
|
C. |
210
|
|
|
|
D. |
210
|
|
|
ITEM 16
|
210
|
||
|
|
A. |
210
|
|
|
|
B. |
211
|
|
|
|
C. |
211
|
|
|
|
D. |
211
|
|
|
|
E. |
211
|
|
|
|
F. |
211
|
|
|
|
G. |
212
|
|
|
|
H. |
212
|
|
|
|
213
|
||
|
ITEM 17.
|
213
|
||
|
ITEM 18.
|
213
|
||
|
ITEM 19.
|
213
|
||
| • |
the initiation, timing, progress and results of our current and future preclinical studies and clinical trials and related preparatory work and the period during which the results of the trials will become available, as well as our research
and development programs;
|
| • |
our estimates regarding expenses, future revenue, capital requirements and needs for additional financing;
|
| • |
our expectations regarding timing of regulatory filings for, or our ability to obtain regulatory approval of, tebentafusp or any of our other product candidates;
|
| • |
our ability to identify and develop additional product candidates using our ImmTAX platform;
|
| • |
business disruptions affecting the initiation, patient enrollment, clinical trial site monitoring, development and operation of our clinical trials, including a public health emergency, such as the ongoing coronavirus 2019, or COVID-19,
pandemic;
|
| • |
the potential benefits of our product candidates;
|
| • |
our expectations regarding the potential commercialization of, the potential market size and the rate and degree of market acceptance for any product candidates that we develop;
|
| • |
our business strategies and goals;
|
| • |
our plans to collaborate, or statements regarding our current collaborations;
|
| • |
our ability to find future partners and collaborators;
|
| • |
the performance of our third-party suppliers and manufacturers,
|
| • |
our expectations regarding our ability to obtain, maintain and enforce intellectual property protection for our product candidates and our ability to operate our business without infringing, misappropriating or otherwise violating the
intellectual property rights of others;
|
| • |
the effects of competition with respect to tebentafusp or any of our other current or future product candidates, as well as innovations by current and future competitors in our industry;
|
| • |
our financial performance and our ability to effectively manage our anticipated growth;
|
| • |
our ability to identify, recruit and retain qualified employees and key personnel;
|
| • |
whether we are classified as a PFIC for current and future periods;
|
| • |
our ability to raise additional capital; and
|
| • |
our estimates regarding future expenses, revenues and needs for additional financing and the accuracy thereof.
|
| Item 1. |
| Item 2. |
| Item 3. |
| A. |
Selected financial data.
|
| B. |
| C. |
| D. |
Risk factors.
|
| • |
We have incurred significant losses in every year since our inception. We expect to continue to incur losses over the next several years and may never achieve or maintain profitability.
|
| • |
We will require substantial additional funding to achieve our business goals. If we are unable to obtain this funding when needed and on acceptable terms, we could be forced to delay, limit or terminate our product development efforts.
|
| • |
We are heavily dependent on the success of our ImmTAX platform to identify and develop product candidates. If we or our collaborators are unable to successfully develop and commercialize our platforms or experience significant delays in
doing so, our business may be harmed.
|
| • |
We may be unable to successfully complete additional large-scale, pivotal clinical trials for any product candidates we develop after tebentafusp.
|
| • |
Our product candidates utilize a novel mechanism of action and involve novel targets which may result in greater research and development expenses, regulatory issues that could delay or prevent approval, or discovery of unknown or
unanticipated adverse effects.
|
| • |
Clinical product development involves a lengthy and expensive process, with an uncertain outcome.
|
| • |
The effects of health epidemics, including the ongoing COVID-19 pandemic, in regions where we, or the third parties on which we rely, have business operations could adversely impact our business, including our pre-clinical studies and
clinical trials, as well as the business or operations of our CROs or other third parties with whom we conduct business.
|
| • |
For a period of six weeks, our IMC-F106C program was put on partial clinical hold in 2020 by the FDA following the death of the second patient dosed in this trial, which was subsequently determined to be unrelated to study drug. The hold has
since been lifted and the trial has been resumed.
|
| • |
We are subject to manufacturing risks that could substantially increase our costs and limit supply of our products.
|
| • |
We face substantial competition, which may result in others developing or commercializing drugs before or more successfully than us.
|
| • |
Our existing collaborations are important to our business, and future collaborations may also be important to us. If we are unable to maintain any of these collaborations, or if these collaborations are not successful, our business could be
adversely affected.
|
| • |
If we are unable to adequately protect our proprietary technology or obtain, maintain, protect and enforce patent and other intellectual property protection for our technology and products or if the scope of the protection obtained is not
sufficiently broad, our competitors and other third parties could develop and commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and products may be impaired.
|
| • |
Third parties may initiate legal proceedings alleging that we are infringing, misappropriating or otherwise violating their intellectual property or proprietary rights, the outcome of which would be uncertain and could have a material
adverse effect on the success of our business.
|
| • |
The FDA regulatory pathways can be difficult to predict and whether, for example, further unanticipated clinical trials are required, will depend on the data obtained in our ongoing clinical trials.
|
| • |
Our future success depends on our ability to retain key executives and experienced scientists and to attract, retain and motivate qualified personnel.
|
| • |
As a company based outside of the United States, we are subject to economic, political, regulatory and other risks associated with international operations.
|
| • |
continue our ongoing and planned development of our five clinical stage programs, including tebentafusp, our lead oncology program, which is being evaluated in a Phase 3 pivotal trial in patients with metastatic uveal melanoma;
|
| • |
initiate pre-clinical studies and clinical trials for any additional product candidates that we may pursue in the future, including our earlier-stage programs;
|
| • |
seek regulatory approvals for tebentafusp and any future product candidates that successfully complete clinical trials;
|
| • |
build a portfolio of product candidates through the discovery, development, or acquisition or in-license of drugs, product candidates or technologies;
|
| • |
establish a sales, marketing, manufacturing and distribution capability to commercialize tebentafusp and any future product candidate for which we may obtain marketing approval;
|
| • |
maintain, protect, enforce and expand our intellectual property portfolio;
|
| • |
acquire or in-license other product candidates, intellectual property and technologies;
|
| • |
hire additional clinical, regulatory and scientific personnel;
|
| • |
add operational, financial and management information systems and personnel, including personnel to support our product development and planned future commercialization efforts; and
|
| • |
incur additional legal, accounting and other expenses associated with operating as a public company.
|
| • |
progress, timing, scope and costs of our clinical trials, including the ability to timely initiate clinical sites, enroll subjects and manufacture soluble bispecific TCR product candidates for our ongoing, planned
and potential future clinical trials, including our Phase 3 clinical trial of tebentafusp in metastatic uveal melanoma, our Phase 1/2 clinical trial of IMC-C103C (MAGE-A4) in multiple solid tumors and our Phase 1/2 clinical trial of IMC-F106C
(PRAME) in multiple solid tumors;
|
| • |
time and costs required to perform research and development to identify and characterize new product candidates from our research programs;
|
| • |
the time and cost necessary to pursue regulatory authorizations and approvals that may be required by regulatory authorities to execute clinical trials or commercialize our products;
|
| • |
our ability to successfully commercialize our product candidates, if approved;
|
| • |
our ability to have clinical and commercial products successfully manufactured consistent with FDA, EMA and other authorities’ regulations;
|
| • |
amount of sales and other revenues from product candidates that we may commercialize, if any, including the selling prices for such potential products and the availability of adequate third-party coverage and reimbursement for patients;
|
| • |
sales and marketing costs associated with commercializing our products, if approved, including the cost and timing of building our marketing and sales capabilities;
|
| • |
cost of building, staffing and validating our manufacturing processes, which may include capital expenditure;
|
| • |
terms and timing of any revenue from our existing collaborations;
|
| • |
costs of operating as a public company;
|
| • |
time and cost necessary to respond to technological, regulatory, political and market developments;
|
| • |
costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights;
|
| • |
costs, associated with, and terms and timing of, any future any potential acquisitions, strategic collaborations, licensing agreements or other arrangements that we may establish; and
|
| • |
inability of clinical sites to enroll patients as healthcare capacities are required to cope with natural disasters, epidemics or other health system emergencies, such as the COVID-19 pandemic.
|
| • |
be delayed in obtaining marketing approval for our product candidates;
|
| • |
not obtain marketing approval at all;
|
| • |
obtain approval for indications or patient populations that are not as broad as intended or desired;
|
| • |
be subject to post-marketing testing requirements; or
|
| • |
have the product removed from the market after obtaining marketing approval.
|
| • |
the research methodology used may not be successful in identifying potential indications and/or product candidates;
|
| • |
potential product candidates may, after further study, be shown to have harmful adverse effects or other characteristics that indicate they are unlikely to be effective products; or
|
| • |
it may take greater human and financial resources than we will possess to identify additional therapeutic opportunities for our product candidates or to develop suitable potential product candidates through internal research programs,
thereby limiting our ability to develop, diversify and expand our product portfolio.
|
| • |
delays or difficulties in enrolling and retaining patients in our clinical trials, including patients that may not be able or willing to comply with clinical trial protocols such as weekly dosing regimens if quarantines impede patient
movement or interrupt healthcare services;
|
| • |
delays or difficulties in clinical site initiation, including difficulties in recruiting and retaining clinical site investigators and clinical site staff;
|
| • |
increased rates of patients withdrawing from our clinical trials following enrollment as a result of risks of exposure to COVID-19, being forced to quarantine or being unable to visit clinical trial locations or otherwise comply with
clinical trial protocols;
|
| • |
diversion or prioritization of healthcare resources away from the conduct of clinical trials and towards the COVID-19 pandemic, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the
conduct of our clinical trials, and because, who, as healthcare providers, may have heightened exposure to COVID-19 and adversely impact our clinical trial operations;
|
| • |
interruption of our clinical supply chain or key clinical trial activities, such as clinical trial site monitoring, due to limitations on travel imposed or recommended by federal, state/provincial or municipal governments, employers and
others; and
|
| • |
limitations in employee resources that would otherwise be focused on the conduct of our clinical trials, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people.
|
| • |
delays in receiving approval from local regulatory authorities to initiate our planned clinical trials;
|
| • |
delays in clinical sites receiving the supplies and materials needed to conduct our clinical trials;
|
| • |
interruption in global shipping that may affect the transport of clinical trial materials, such as investigational drug product and comparator drugs used in our clinical trials;
|
| • |
changes in federal, state/provincial or municipal regulations as part of a response to the COVID-19 coronavirus outbreak which may require us to change the ways in which our clinical trials are conducted, which may result in unexpected
costs, or to discontinue the clinical trials altogether;
|
| • |
delays in necessary interactions with local regulators, ethics committees and other important agencies and contractors due to limitations in employee resources or forced furlough of government employees; and
|
| • |
the refusal of the FDA to accept data from clinical trials in these affected geographies.
|
| • |
the FDA, EMA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials;
|
| • |
the FDA, EMA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials;
|
| • |
we may be unable to demonstrate to the satisfaction of the FDA, EMA or comparable foreign regulatory authorities that a product candidate is safe and effective for its proposed indication or a related companion diagnostic is suitable to
identify appropriate patient populations;
|
| • |
the results of clinical trials may not meet the level of statistical significance required by the FDA, EMA or comparable foreign regulatory authorities for approval;
|
| • |
we may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks;
|
| • |
the FDA, EMA or comparable foreign regulatory authorities may disagree with our interpretation of data from pre-clinical studies or clinical trials;
|
| • |
the data collected from clinical trials of our product candidates may not be sufficient to support the submission a BLA or other submission or to obtain regulatory approval in the United States or elsewhere;
|
| • |
the FDA, EMA or comparable foreign regulatory authorities may find deficiencies with or fail to approve the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; and
|
| • |
the approval policies or regulations of the FDA, EMA or comparable foreign regulatory authorities may significantly change such that our clinical data are insufficient for approval.
|
| • |
regulators or institutional review boards, or IRBs, or ethics committees may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site;
|
| • |
we may experience delays in reaching, or fail to reach, agreement on acceptable terms with prospective trial sites and prospective CROs, the terms of which can be subject to extensive negotiation and may vary significantly among different
CROs and trial sites;
|
| • |
clinical trials of our product candidates may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional pre-clinical studies or clinical trials or we may decide to abandon product
development programs;
|
| • |
the number of patients required for clinical trials of our product candidates may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate or participants may drop out of these clinical trials or
fail to return for post-treatment follow-up at a higher rate than we anticipate;
|
| • |
our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all, or may deviate from the clinical trial protocol or drop out of the trial, which may
require that we add new clinical trial sites or investigators;
|
| • |
we may elect to, or regulators or IRBs or ethics committees may require us or our investigators to, suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or a finding that the
participants are being exposed to unacceptable health risks;
|
| • |
the cost of clinical trials of our product candidates may be greater than we anticipate;
|
| • |
the supply or quality of our product candidates or other materials necessary to conduct clinical trials of our product candidates may be insufficient or inadequate; and
|
| • |
our product candidates may have undesirable side effects or other unexpected characteristics, causing us or our investigators, regulators or IRBs or ethics committees to suspend or terminate the trials, or reports may arise from pre-clinical
or clinical testing of other cancer therapies that raise safety or efficacy concerns about our product candidates.
|
| • |
the severity of the disease under investigation;
|
| • |
the eligibility criteria for the clinical trial in question;
|
| • |
the availability of an appropriate genomic screening test;
|
| • |
the perceived risks and benefits of the product candidate under study;
|
| • |
the efforts to facilitate timely enrollment in clinical trials;
|
| • |
the patient referral practices of physicians;
|
| • |
the ability to monitor patients adequately during and after treatment;
|
| • |
the proximity and availability of clinical trial sites for prospective patients; and
|
| • |
factors we may not be able to control, such as current or potential pandemics that may limit patients, principal investigators or staff or clinical site availability (e.g., outbreak of COVID-19).
|
| • |
our available capital resources or capital constraints we experience;
|
| • |
the rate of progress, costs, and results of our clinical trials and research and development activities, including the extent of scheduling conflicts with participating clinicians and collaborators;
|
| • |
our ability to identify and enroll patients who meet clinical trial eligibility criteria;
|
| • |
our receipt of approvals by the FDA, EMA and comparable foreign regulatory authorities, and the timing thereof;
|
| • |
other actions, decisions, or rules issued by regulators;
|
| • |
our ability to access sufficient, reliable, and affordable supplies of materials used in the manufacture of our product candidates;
|
| • |
our ability to manufacture and supply clinical trial materials to our clinical sites on a timely basis;
|
| • |
the efforts of our collaborators with respect to the commercialization of our approved products, if any; and
|
| • |
the securing of, costs related to, and timing issues associated with, commercial product manufacturing, as well as sales and marketing activities.
|
| • |
differing regulatory requirements in foreign countries;
|
| • |
unexpected changes in tariffs, trade barriers, price and exchange controls and other regulatory requirements;
|
| • |
differing standards for the conduct of clinical trials;
|
| • |
increased difficulties in managing the logistics and transportation of storing and shipping product candidates produced in the United States or elsewhere and shipping the product candidate to patients in other countries;
|
| • |
import and export requirements and restrictions;
|
| • |
economic weakness, including inflation, or political instability in foreign economies and markets;
|
| • |
compliance with tax, employment, immigration and labor laws for employees living or traveling abroad;
|
| • |
foreign taxes, including withholding of payroll taxes;
|
| • |
foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country;
|
| • |
difficulties staffing and managing foreign operations;
|
| • |
workforce uncertainty in countries where labor unrest is more common than in the United Kingdom or the United States;
|
| • |
differing payor reimbursement regimes, governmental payors or patient self-pay systems, and price controls;
|
| • |
potential liability under the Foreign Corrupt Practices Act of 1977, as amended, the U.K. Bribery Act 2010, or comparable foreign regulations;
|
| • |
challenges enforcing or protecting our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States or the United
Kingdom;
|
| • |
the impacts Brexit may have with respect to the cross-border acknowledgment of clinical trial results and marketing authorizations as well as recruitment of scientific personnel;
|
| • |
production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and
|
| • |
business interruptions resulting from geo-political actions, including war and terrorism.
|
| • |
the inability to recruit, train and retain adequate numbers of effective sales and marketing personnel;
|
| • |
the inability of sales personnel to obtain access to physicians or persuade adequate numbers of physicians to prescribe any future product that we may develop;
|
| • |
the lack of complementary treatments to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and
|
| • |
unforeseen costs and expenses associated with creating an independent sales and marketing organization.
|
| • |
the clinical indications for which our product candidates are approved;
|
| • |
physicians, hospitals, cancer treatment centers, and patients considering our product candidates as a safe and effective treatment;
|
| • |
hospitals and cancer treatment centers establishing the infrastructure required for the administration of the product candidate;
|
| • |
the potential and perceived advantages of our product candidates over alternative treatments;
|
| • |
the prevalence and severity of any side effects;
|
| • |
product labeling or product insert requirements of the FDA, the EMA or other regulatory authorities;
|
| • |
limitations or warnings contained in the labeling approved by the FDA or the EMA;
|
| • |
the timing of market introduction of our product candidates as well as competitive products;
|
| • |
the cost of treatment in relation to alternative treatments;
|
| • |
the amount of upfront costs or training required for physicians to administer our product candidates;
|
| • |
the pricing of our products and the availability of coverage and adequate reimbursement by third-party payors and government authorities;
|
| • |
the willingness of patients to pay out-of-pocket in the absence of comprehensive coverage and adequate reimbursement by third-party payors and government authorities;
|
| • |
relative convenience and ease of administration, including as compared to alternative treatments and competitive therapies; and
|
| • |
the effectiveness of our sales and marketing efforts and distribution support.
|
| • |
the impairment of our business reputation;
|
| • |
the withdrawal of clinical trial participants;
|
| • |
costs due to related litigation;
|
| • |
the distraction of management’s attention from our primary business;
|
| • |
substantial monetary awards to patients or other claimants;
|
| • |
the inability to commercialize our product candidates; and
|
| • |
decreased demand for our product candidates, if approved for commercial sale.
|
| • |
collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations;
|
| • |
collaborators may not perform their obligations as expected;
|
| • |
collaborators may not pursue development and commercialization of any product candidates that achieve regulatory approval or may elect not to continue or renew development or commercialization programs based on clinical trial results,
changes in the collaborators’ strategic focus or available funding, or external factors, such as an acquisition, that divert resources or create competing priorities;
|
| • |
collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product
candidate for clinical testing;
|
| • |
collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products or product candidates if the collaborators believe that competitive products are more likely to be
successfully developed or can be commercialized under terms that are more economically attractive than ours; this may also happen if the collaborators’ development of competing products is substantially faster than our development timelines;
|
| • |
collaborators may not further develop product candidates developed by us or co-developed with us under the collaboration;
|
| • |
product candidates discovered in collaboration with us may be viewed by our collaborators as competitive with their own product candidates or products, which may cause collaborators to cease to devote resources to the commercialization of
our product candidates;
|
| • |
a collaborator with marketing and distribution rights to one or more of our product candidates that achieve regulatory approval may not commit sufficient resources to the marketing and distribution of such product or products;
|
| • |
disagreements with collaborators, including disagreements over proprietary rights, contract interpretation or the preferred course of development, might cause delays or termination of the research, development or commercialization of product
candidates, might lead to additional responsibilities for us with respect to product candidates, or might result in litigation or arbitration, any of which would be time-consuming and expensive;
|
| • |
collaborators have certain defined rights to change or expand the scope of development programs during the course of the collaboration. This may lead to additional research work for us that may be time-consuming and expensive. Such work may
compete with our own development programs and may delay timelines to market or proof-of-concept for our product candidates. If development programs under the collaboration turn out to be more costly and time-consuming, such unanticipated costs
and work could likewise compete with our internal development programs;
|
| • |
collaborators may not properly maintain, enforce or defend our intellectual property or proprietary information or may use them in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or
proprietary information or expose us to potential litigation;
|
| • |
collaborators may infringe, misappropriate or otherwise violate the intellectual property or proprietary rights of third parties, which may expose us to litigation and potential liability, and collaborators may also allege that we are liable
for potential infringement, misappropriation or other violations of third-party intellectual property or proprietary rights during the research and development work for the collaboration;
|
| • |
certain collaborations may be terminated for the convenience of the collaborator and, if terminated, we could be required to raise additional capital to pursue further development or commercialization of the applicable product candidates.
For example, certain of our collaboration and license agreements may be terminated for convenience upon the completion of a specified notice period; and
|
| • |
collaborators may discontinue the development of product candidates within the collaboration, for example if they consider the results achieved so far or the product candidates not promising enough or if their development strategies change.
|
| • |
have staffing difficulties;
|
| • |
fail to comply with contractual obligations;
|
| • |
experience regulatory compliance issues;
|
| • |
undergo changes in priorities or become financially distressed; or
|
| • |
form relationships with other entities, some of which may be our competitors.
|
| • |
reliance on the third party for regulatory compliance and quality assurance;
|
| • |
the possible breach of the manufacturing agreement by the third party;
|
| • |
the possible misappropriation or unauthorized disclosure of our proprietary information, including our trade secrets and know-how; and
|
| • |
the possible termination or nonrenewal of the agreement by the third party at a time that is costly or inconvenient for us.
|
| • |
the scope of rights granted under the agreement and other interpretation-related issues;
|
| • |
the extent to which our technology and processes infringe on intellectual property of the counterparty that is not subject to the agreement;
|
| • |
the sublicensing of patent and other intellectual or proprietary rights under our collaborative development relationships;
|
| • |
our diligence obligations under the agreement and what activities satisfy those diligence obligations;
|
| • |
the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our counterparty and us and our partners; and
|
| • |
the priority of invention of patented technology.
|
| • |
others may be able to make products that are similar to our product candidates or utilize similar technology but that are not covered by the claims of the patents that we own or license now or in the future;
|
| • |
we or our licensors or collaborators might not have been the first to make the inventions covered by the issued patents or pending patent applications that we own or license now or in the future;
|
| • |
we or our licensors or collaborators might not have been the first to file patent applications covering certain of our or their inventions;
|
| • |
others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights or any intellectual property rights we may license;
|
| • |
it is possible that our present or future pending patent applications (whether owned or licensed) will not lead to issued patents;
|
| • |
it is possible that there are or will be prior public disclosures that could invalidate our or our licensors’ or collaboration partners’ patents;
|
| • |
issued patents that we hold rights to may fail to provide us with any competitive advantage, or may be held invalid or unenforceable, including as a result of legal challenges by our competitors or other third parties;
|
| • |
our competitors or other third parties might conduct research and development activities in countries where we do not have patent rights or in countries where research and development safe harbor laws exist, and then use the information
learned from such activities to develop competitive products for sale in our major commercial markets;
|
| • |
we may not develop additional proprietary technologies that are patentable;
|
| • |
the ownership, validity or enforceability of our patents or patent applications may be challenged by third parties;
|
| • |
the patents or pending or future applications of others, if issued, may harm our business; and
|
| • |
we may choose not to file a patent in order to maintain certain trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property.
|
| • |
the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our or our collaborators’ clinical trials;
|
| • |
we or our collaborators may be unable to demonstrate to the satisfaction of the FDA or comparable foreign regulatory authorities that our product candidates are safe, pure, potent and have a favorable risk/benefit profile for any of their
proposed indications;
|
| • |
the results of clinical trials may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval;
|
| • |
the FDA or comparable foreign regulatory authorities may disagree with our interpretation of data from pre-clinical programs or clinical trials;
|
| • |
data collected from clinical trials of product candidates may not be sufficient to the satisfaction of the FDA or comparable foreign regulatory authorities to support the submission of a BLA or other comparable submission in foreign
jurisdictions or to obtain regulatory approval in the United States or elsewhere;
|
| • |
manufacturing processes or facilities or those of the third-party manufacturers we use may not be adequate to support approval of our product candidates; and
|
| • |
the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval.
|
| • |
restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls;
|
| • |
clinical trial holds;
|
| • |
fines, warning letters or other regulatory enforcement action;
|
| • |
refusal by the FDA to approve pending applications or supplements to approved applications filed by us;
|
| • |
product seizure or detention, or refusal to permit the import or export of products; and
|
| • |
injunctions or the imposition of civil or criminal penalties.
|
| • |
the demand for our current or future product candidates, if we obtain regulatory approval;
|
| • |
our ability to set a price that we believe is fair for our products;
|
| • |
our ability to obtain coverage and adequate reimbursement for a product;
|
| • |
our ability to generate revenue and achieve or maintain profitability;
|
| • |
the level of taxes that we are required to pay; and
|
| • |
the availability of capital.
|
| • |
the federal Anti-Kickback Statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the
referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under federal and state healthcare programs such as Medicare and Medicaid. A person or entity does not need to have
actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
|
| • |
the federal civil and criminal false claims and civil monetary penalties laws, including the federal False Claims Act, or FCA, imposes criminal and civil penalties, including through civil whistleblower or qui tam actions, against
individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the
federal government. In addition, the government may assert that a claim including items and services resulting from a violation of the federal Anti-Kickback Statute constitutes a false of fraudulent claim for purposes of the False Claims Act;
|
| • |
the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or
covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services; similar to the federal Anti-Kickback Statute, a person or entity does not need to
have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
|
| • |
the federal physician payment transparency requirements, sometimes referred to as the “Sunshine Act” under the Affordable Care Act, require manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare,
Medicaid, or the Children’s Health Insurance Program to report to CMS information related to transfers of value made to physicians (currently defined to include doctors, dentists, optometrists, podiatrists and
chiropractors) and teaching hospitals, as well as ownership and investment interests of such physicians and their immediate family members. Effective January 1, 2022, these reporting obligations will extend to include transfers of value made
to certain non-physician providers including physician assistants, nurse practitioners, clinical nurse specialists, anesthesiologist assistants, certified registered nurse anesthetists and certified nurse midwives during the previous year;
|
| • |
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 and its implementing regulations, impose obligations on certain covered entity healthcare providers, health plans, and healthcare
clearinghouses as well as their business associates that perform certain services involving the use or disclosure of individually identifiable health information and their subcontractors that use, disclose or otherwise process individually
identifiable health information, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; and
|
| • |
analogous state laws and regulations, such as state anti-kickback and false claims laws may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors,
including private insurers. Some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to
requiring drug manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures. Further, many state laws governing the privacy and security of health information in certain
circumstances, differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
|
| • |
convey, sell, lease, transfer, assign, dispose of or otherwise make cash payments consisting of all or any part of our business or property;
|
| • |
effect certain changes in our business, management, ownership or business locations;
|
| • |
merge or consolidate with, or acquire all or substantially all of the capital stock or assets of, any other company;
|
| • |
create, incur, assume or be liable for any additional indebtedness, or create, incur, allow or permit to exist any additional liens;
|
| • |
pay cash dividends on, make any other distributions in respect of, or redeem, retire or repurchase, any shares of our capital stock;
|
| • |
make certain investments; and
|
| • |
enter into transactions with our affiliates.
|
| • |
increased operating expenses and cash requirements;
|
| • |
the assumption of additional indebtedness or contingent liabilities;
|
| • |
assimilation of operations, intellectual property and products of an acquired company or product, including difficulties associated with integrating new personnel;
|
| • |
the diversion of our management’s attention from our existing product programs and initiatives in pursuing such a strategic merger or acquisition;
|
| • |
retention of key employees, the loss of key personnel, and uncertainties in our ability to maintain key business relationships;
|
| • |
risks and uncertainties associated with the other party to such a transaction, including the prospects of that party and their existing products or product candidates and regulatory approvals; and
|
| • |
our inability to generate revenue from acquired technology and/or products sufficient to meet our objectives in undertaking the acquisition or even to offset the associated acquisition and maintenance costs.
|
| • |
economic weakness, including inflation, or political instability in particular non-U.S. economies and markets;
|
| • |
differing and changing regulatory requirements for product approvals;
|
| • |
differing jurisdictions could present different issues for securing, maintaining or obtaining freedom to operate in such jurisdictions;
|
| • |
potentially reduced protection for intellectual property and proprietary rights;
|
| • |
difficulties in compliance with different, complex and changing laws, regulations and court systems of multiple jurisdictions and compliance with a wide variety of foreign laws, treaties and regulations;
|
| • |
changes in non-U.S. regulations and customs, tariffs and trade barriers;
|
| • |
changes in non-U.S. currency exchange rates of the pound sterling, U.S. dollar, euro and currency controls;
|
| • |
changes in a specific country’s or region’s political or economic environment, including the implications of the recent decision of the United Kingdom to withdraw from the European Union;
|
| • |
trade protection measures, import or export licensing requirements or other restrictive actions by governments;
|
| • |
differing reimbursement regimes and price controls in certain non-U.S. markets;
|
| • |
negative consequences from changes in tax laws;
|
| • |
compliance with tax, employment, immigration and labor laws for employees living or traveling abroad, including, for example, the variable tax treatment in different jurisdictions of options granted under our share option schemes or equity
incentive plans;
|
| • |
workforce uncertainty in countries where labor unrest is more common than in the United States;
|
| • |
litigation or administrative actions resulting from claims against us by current or former employees or consultants individually or as part of class actions, including claims of wrongful terminations, discrimination, misclassification or
other violations of labor law or other alleged conduct;
|
| • |
difficulties associated with staffing and managing international operations, including differing labor relations;
|
| • |
production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and
|
| • |
business interruptions resulting from geo-political actions, including war and terrorism, or natural disasters including earthquakes, typhoons, floods and fires.
|
| • |
the commencement, enrollment or results of our planned and future clinical trials;
|
| • |
adverse results or delays in pre-clinical studies or clinical trials;
|
| • |
reports of adverse events in products similar or perceived to be similar to those we are developing or clinical trials of such products;
|
| • |
an inability to obtain additional funding;
|
| • |
failure by us to successfully develop and commercialize our product candidates;
|
| • |
failure by us to maintain our existing strategic collaborations or enter into new collaborations;
|
| • |
failure by us to identify additional product candidates for our pipeline;
|
| • |
failure by us or our licensors and strategic partners to prosecute, maintain, protect or enforce our intellectual property and proprietary rights;
|
| • |
disputes or other developments relating to intellectual and other proprietary rights, including litigation
|
| • |
matters and our ability to obtain patent and other intellectual property protection for our technologies;
|
| • |
changes in laws or regulations applicable to future products;
|
| • |
an inability to obtain adequate product supply for our product candidates or the inability to do so at acceptable prices;
|
| • |
adverse regulatory decisions;
|
| • |
the introduction of new products, services or technologies by our competitors;
|
| • |
failure by us to meet or exceed financial projections we may provide to the public;
|
| • |
failure by us to meet or exceed the financial projections of the investment community;
|
| • |
the perception of the pharmaceutical industry by the public, legislatures, regulators and the investment community;
|
| • |
changes in the structure of healthcare payment systems;
|
| • |
announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us, our strategic partner or our competitors;
|
| • |
additions or departures of key scientific or management personnel;
|
| • |
significant lawsuits, including patent or shareholder litigation;
|
| • |
changes in the market valuations of similar companies;
|
| • |
general economic, industry, political and market conditions, including, but not limited to, the ongoing impact of the COVID-19 pandemic;
|
| • |
sales of our ADSs or ordinary shares by us or our shareholders in the future; and
|
| • |
the trading volume of our ADSs.
|
| • |
delaying, deferring, or preventing a change in control;
|
| • |
entrenching our management and/or the board of directors;
|
| • |
impeding a merger, scheme of arrangement, takeover, or other business combination involving us; or
|
| • |
discouraging a potential acquirer from making a takeover offer or otherwise attempting to obtain control of us.
|
| • |
not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, or Section 404;
|
| • |
not being required to comply with any requirement that has or may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information
about the audit and the financial statements;
|
| • |
being permitted to provide only two years of audited financial statements in this initial registration statement, in addition to any required unaudited interim financial statements, with correspondingly reduced “Management’s Discussion and
Analysis of Financial Condition and Results of Operations” disclosure;
|
| • |
reduced disclosure obligations regarding executive compensation; and
|
| • |
an exemption from the requirement to seek nonbinding advisory votes on executive compensation or golden parachute arrangements.
|
| • |
In connection with a potential offer, if following an approach by or on behalf of a potential bidder, the company is “the subject of rumor or speculation” or there is an “untoward movement” in the company’s share price, there is a
requirement for the potential bidder to make a public announcement about a potential offer for the company, or for the company to make a public announcement about its review of a potential offer.
|
| • |
When a person or group of persons acting in concert (a) acquires, whether by a series of transactions over a period of time or not, interests in shares carrying 30% or more of the voting rights of a company (which percentage is treated by
the Takeover Code as the level at which effective control is obtained) or (b) acquires an interest in any other shares which increases the percentage of shares carrying voting rights in which they are interested when they are already interested
in shares which carry not less than 30% of the voting rights but do not hold shares carrying more than 50% of such voting rights, they must make a cash offer to all other shareholders at the highest price paid by them or any person acting in
concert with them in the 12 months before the offer was announced.
|
| • |
When interests in shares carrying 10% or more of the voting rights of a class have been acquired by an offeror (i.e., a bidder) and any person acting in concert with it in the offer period (i.e., before the shares subject to the offer have
been acquired) or within the previous 12 months, the offer must be in cash or be accompanied by a cash alternative for all shareholders of that class at the highest price paid by the offeror or any person acting in concert with them in that
period. Further, if an offeror or any person acting in concert with them acquires any interest in shares during the offer period, the offer for the shares must be in cash or accompanied by a cash alternative at a price at least equal to the
price paid for such shares during the offer period.
|
| • |
If after an announcement is made, the offeror or any person acting in concert with them acquires an interest in shares in an offeree company (i.e., a target) at a price higher than the value of the offer, the offer must be increased to not
less than the highest price paid for the interest in shares so acquired.
|
| • |
The board of directors of the offeree company must appoint a competent independent adviser whose advice on the financial terms of the offer must be made known to all the shareholders, together with the opinion of the board of directors of
the offeree company.
|
| • |
Special or favorable deals for selected shareholders are not permitted, except in certain circumstances where independent shareholder approval is given and the arrangements are regarded as fair and reasonable in the opinion of the financial
adviser to the offeree.
|
| • |
All shareholders must be given the same information.
|
| • |
Each document published in connection with an offer by or on behalf of the offeror or offeree must state that the directors of the offeror or the offeree, as the case may be, accept responsibility for the information contained therein.
|
| • |
Profit forecasts, quantified financial benefits statements and asset valuations must be made to specified standards and must be reported on by professional advisers.
|
| • |
Misleading, inaccurate or unsubstantiated statements made in documents or to the media must be publicly corrected immediately.
|
| • |
Actions during the course of an offer by the offeree company, which might frustrate the offer are generally prohibited unless shareholders approve these plans. Frustrating actions would include, for example, lengthening the notice period for
directors under their service contract or agreeing to sell off material parts of the target group.
|
| • |
Stringent and detailed requirements are laid down for the disclosure of dealings in relevant securities during an offer, including the prompt disclosure of positions and dealing in relevant securities by the parties to an offer and any
person who is interested (directly or indirectly) in 1% or more of any class of relevant securities.
|
| • |
Employees of both the offeror and the offeree company and the trustees of the offeree company’s pension scheme must be informed about an offer. In addition, the offeree company’s employee representatives and pension scheme trustees have the
right to have a separate opinion on the effects of the offer on employment appended to the offeree board of directors’ circular or published on a website.
|
| • |
under our articles of association, any resolution put to the vote of a general meeting must be decided exclusively on a poll. Under English law, it would be possible for our articles of association to be
amended such that each shareholder present at a meeting has only one vote unless demand is made for a vote on a poll, in which case each holder gets one vote per share owned. Under U.S. law, each shareholder typically is entitled to one vote
per share at all meetings;
|
| • |
under English law, subject to certain exceptions and disapplications, each shareholder generally has preemptive rights to subscribe on a proportionate basis to any issuance of ordinary shares or rights to subscribe for, or to convert
securities into, ordinary shares for cash. Under U.S. law, shareholders generally do not have preemptive rights unless specifically granted in the certificate of incorporation or otherwise;
|
| • |
under English law and our articles of association, certain matters require the approval of 75% of the shareholders who vote (in person or by proxy) on the relevant resolution (or on a poll of shareholders representing 75% of the ordinary
shares voting (in person or by proxy)), including amendments to the articles of association. This may make it more difficult for us to complete corporate transactions deemed advisable by our board of directors. Under U.S. law, generally only
majority shareholder approval is required to amend the certificate of incorporation or to approve other significant transactions;
|
| • |
in the United Kingdom, takeovers may be structured as takeover offers or as schemes of arrangement. Under English law, a bidder seeking to acquire us by means of a takeover offer would need to make an offer for all of our outstanding
ordinary shares/ADSs. If acceptances are not received for 90% or more of the ordinary shares/ADSs under the offer, under English law, the bidder cannot complete a “squeeze out” to obtain 100% control of us. Accordingly, acceptances of 90% of
our outstanding ordinary shares/ADSs will likely be a condition in any takeover offer to acquire us, not 50% as is more common in tender offers for corporations organized under Delaware law. By contrast, a scheme of arrangement, the successful
completion of which would result in a bidder obtaining 100% control of us, requires the approval of a majority of shareholders voting at the meeting and representing 75% of the ordinary shares voting for approval;
|
| • |
under English law and our articles of association, shareholders and other persons whom we know or have reasonable cause to believe are, or have been, interested in our shares may be required to disclose information regarding their interests
in our shares upon our request, and the failure to provide the required information could result in the loss or restriction of rights attaching to the shares, including prohibitions on certain transfers of the shares, withholding of dividends
and loss of voting rights. Comparable provisions generally do not exist under U.S. law; and
|
| • |
the quorum requirement for a shareholders’ meeting is a minimum of two shareholders entitled to vote at the meeting and present in person or by proxy or, in the case of a shareholder which is a corporation, represented by a duly authorized
representative. Under U.S. law, a majority of the shares eligible to vote must generally be present (in person or by proxy) at a shareholders’ meeting in order to constitute a quorum. The minimum number of shares required for a quorum can be
reduced pursuant to a provision in a company’s certificate of incorporation or bylaws, but typically not below one-third of the shares entitled to vote at the meeting.
|
| A. |
| B. |
Business overview.
|
| • |
Tebentafusp, our ImmTAC molecule targeting an HLA-A*02:01 gp100 antigen, demonstrated monotherapy activity and recently achieved the primary endpoint of superior overall
survival at the first pre-planned interim analysis of a randomized Phase 3 clinical trial in patients with previously untreated metastatic uveal melanoma. We anticipate completing submission of a BLA to the FDA in the third quarter of 2021,
followed by a Marketing Authorization Application, or MAA, submission to the European Medicines Agency, or EMA.
|
| • |
IMC-C103C, our ImmTAC molecule targeting an HLA-A*02:01 MAGE-A4 antigen, is currently being evaluated in a first-in-human Phase 1/2 dose escalation trial in patients with solid
tumor cancers including non-small-cell lung cancer, or NSCLC, gastric, head and neck, ovarian and synovial sarcoma. We are developing this program under a co-development collaboration with Genentech, Inc., or Genentech, under which we have an
option to retain 50% of the economics. We anticipate reporting Phase 1 initial data from this trial in the second half of 2021.
|
| • |
IMC-F106C, our ImmTAC molecule targeting an optimal HLA-A*02:01 PRAME antigen identified with our ImmSPECT target identification platform, is currently being evaluated in a
first-in-human, Phase 1/2 dose escalation trial in patients with multiple solid tumor cancers. PRAME is overexpressed in many solid tumors, including NSCLC, SCLC, endometrial, ovarian, and breast cancers. We anticipate reporting Phase 1 initial
data from this trial in mid-2022.
|
| • |
IMC-I109V, our ImmTAV molecule targeting a conserved HBV envelope antigen, is our most advanced ImmTAV program and is currently being evaluated in a Phase 1/2 clinical trial in
patients with chronic HBV who are non-cirrhotic, hepatitis B e-Antigen negative, and virally suppressed on chronic nucleot(s)ide analogue therapy. Our goal is to develop a functional cure for HBV and we anticipate commencing dosing in our Phase
1 single ascending dose, or SAD, trial in mid-2021. We are also developing a next-generation version of this molecule leveraging our research into universal HLA-E molecules which could benefit a much larger patient population as compared to
classical-HLA antigens.
|
| • |
IMC-M113V, our ImmTAV molecule targeting an HIV gag antigen bispecific TCR molecule, is currently in pre-clinical development. Our HIV programs are funded by the Bill &
Melinda Gates Foundation, or the Gates Foundation, and we are required to make any successfully approved products available at reduced prices in certain developing countries. We retain full development and commercial right in non-developing
countries.
|
| • |
Secure marketing approval for, and then commercialize, tebentafusp, our lead ImmTAC, for the treatment of metastatic uveal melanoma. Our most advanced oncology therapeutic
candidate, tebentafusp, has demonstrated superior overall survival benefit as a monotherapy in a randomized Phase 3 clinical trial in previously untreated metastatic uveal melanoma, a cancer that has historically proven to be insensitive to
other immunotherapies. This primary endpoint was achieved with a hazard ratio of 0.51 (95% CI: 0.36, 0.71; p< 0.0001) at the first pre-planned interim analysis. We intend to seek regulatory approval for tebentafusp in the United States and
Europe. We believe achieving regulatory approval of tebentafusp would provide validation of our entire ImmTAX platform. If tebentafusp is approved, we also believe it will present us with an attractive commercial opportunity, which we intend to
pursue using a targeted commercialization strategy that requires minimal internal infrastructure.
|
| • |
Advance our IMC-C103C program targeting MAGE-A4 for the treatment of solid tumors in collaboration with Genentech. We believe IMC-C103C has the potential to treat a wide range
of solid tumors, including NSCLC. We are currently evaluating IMC-C103C in a first-in-human, Phase 1/2 dose escalation trial in patients with solid tumor cancers. We are developing this program under a co-development collaboration with
Genentech, and are jointly progressing clinical development of IMC-C103C with a partner who possesses deep expertise in clinical development and regulatory strategy. We anticipate reporting Phase 1 initial data from this trial in the second
half of 2021.
|
| • |
Advance our IMC-F106C program targeting PRAME for the treatment of solid tumors. IMC-F106C represents a significant commercial opportunity given the prevalence of the PRAME
target across various cancers. PRAME is overexpressed in many solid tumors, including NSCLC, SCLC, endometrial, ovarian, esophageal, head and neck squamous cell carcinoma, and urothelial cancers. PRAME is also overexpressed in some
hematological malignancies, including acute myeloid leukemia. PRAME expression is generally identified as a poor prognostic feature. We are currently evaluating IMC-F106C in a first-in-human, Phase 1/2 dose escalation trial in patients with
solid tumor cancers including NSCLC, gastric, head and neck, ovarian and synovial sarcoma. We anticipate reporting Phase 1 initial data from this trial in mid-2022.
|
| • |
Advance our IMC-I109V program for the treatment of chronic HBV. Current standard-of-care antiviral agents for HBV do not provide a permanent cure in most cases. Therefore,
lifelong treatment is necessary to lower the risk of chronic HBV-related complications and there remains a large unmet need for a functional cure. The goal of our IMC-I109V program is to develop a functional cure for chronic HBV. If successful,
we believe our therapeutic will allow patients to have a finite period of treatment that will also reduce the risks of end-stage liver disease and hepatocellular carcinoma, which are not completely eliminated by currently available treatments.
We have begun screening patients for our first-in-human, Phase 1/2 clinical trial of IMC-I109V and anticipate commencing dosing in our Phase 1 SAD trial in mid-2021.
|
| • |
Continue to develop our novel universal ImmTAX platform to meaningfully broaden the eligible patient pool. We are developing universal TCR therapeutics that are designed to be
unrestricted by classical HLA status, which would have the potential to significantly increase the patient pool eligible for our therapeutics. Having pioneered the engineering of TCR bispecifics against classical HLA targets, we believe we are
now at the forefront of ushering in a new era of TCR therapies by unlocking universal HLAs, such as HLA-E. This new approach, which we have validated in pre-clinical studies, offers the potential for all patients globally to benefit from a
single therapeutic per target rather than requiring several classical HLA programs with their associated development costs.
|
| • |
Continue to invest in our platform to discover and develop novel therapeutics. To remain an industry leader in TCR bispecifics, we intend to continue identifying and validating
unique targets as well as optimizing current TCRs to continue to improve outcomes for patients across a broad range of diseases.
|
| • |
Opportunistically pursue strategic partnerships to maximize the full potential of our pipeline and ImmTAX platform. We intend to selectively evaluate partnerships to explore
combination therapies and access our partners’ industry-leading capabilities. We plan to assess opportunities to partner with large pharmaceutical companies in the areas of infectious disease and autoimmune diseases to access a broad commercial
infrastructure for those indications.
|

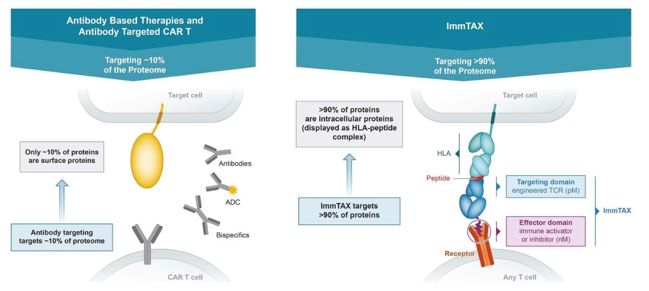
| • |
ImmTAC - Immune mobilizing monoclonal TCRs
Against Cancer
|
| • |
ImmTAV - Immune mobilizing monoclonal TCRs
Against Viruses
|
| • |
ImmTAAI - Immune modulating monoclonal TCRs
Against AutoImmune disease
|



| • |
Phase 1 first-in-human clinical trial (n=84) demonstrated monotherapy activity per RECIST and immune related responses in uveal and cutaneous melanoma patients.
|
| • |
Phase 2 clinical trial (n=127) demonstrated improved overall survival in a cross-trial comparison to a recent metanalysis based on prior clinical trials in a similar previously treated uveal melanoma patient
population (n=287). The cross trial overall survival hazard ratio was 0.50 (95% CI 0.38,0.66).
|
| • |
Phase 3 randomized clinical trial (n=378) achieved the primary endpoint of superior overall survival in the intent-to-treat population with a hazard ratio of 0.51 (95% CI: 0.36, 0.71), p<0.0001 favoring tebentafusp
over investigators choice.
|

| • |
Phase 1 portion (dose escalation): This portion of the clinical trial defined the intra-patient dose escalation regimen, with a top dose of 68 mcg, which was then advanced as
the recommended dose in the Phase 2 portion of the trial as well as our ongoing Phase 3 clinical trial. Of the 19 patients in the Phase 1 portion, we observed three patients had tumor responses that met the criteria defined by RECIST. An
additional four patients did not meet RECIST criteria but had immune-related responses, a category of response previously described for the immune checkpoint therapies, and also suggested improved survival results that supported further
studies.
|
| • |
Phase 2 portion (expansion): This portion of the clinical trial was intended to evaluate the efficacy of tebentafusp in 127 patients with metastatic uveal melanoma as a
second-line or later treatment. The primary endpoint was to estimate the objective response rate, or ORR, under RECIST 1.1 according to an independent central review committee. We believe the observation of immune-related responses in the Phase
1 portion of the trial and the Phase 1 first-in-human trial indicates that overall survival, which captures benefit from RECIST and immune related responses, is a better measurement of treatment effect for tebentafusp, and thus, we included
observation of immune-related responses as a secondary endpoint of this trial. Of the 127 metastatic uveal melanoma patients treated, all had received prior treatments and the majority had received prior immunotherapy regimens (73.2% had prior
immunotherapy; 65.4% had prior anti-PD-1).
|





|
a.
|
Investigator assignment of causality;
|
| b. |
LFT elevation and rash are composites of preferred terms;
|
| c. |
CRS assessed retrospectively by ASTCT (Lee 2019) criteria
|




| • |
HLA-E target identification and validation: HLA-E peptide antigens are significantly more unstable than classical HLA peptide complexes and fall apart within minutes rather than
hours. Therefore, we have developed a suite of four new HLA-E target identification and validation assays that have allowed us to identify novel HLA-E targets for HBV, HIV, TB and a number of oncology targets.
|
| • |
Antigen stabilization: HLA-E/peptide instability also makes the isolation and engineering of specific TCRs challenging. We developed and patented a new HLA-E stabilization
approach that allows highly specific TCRs to be isolated and engineered.
|
| • |
Sufficiently high specificity: HLA-E presents peptides that tend to have a high degree of similarity in their sequence, making it challenging to introduce sufficient levels of
specificity to support clinical development. We have successfully adapted existing specificity tools to overcome these challenges.
|
| • |
Biovian Ltd., headquartered in Turku, Finland, for early-phase clinical drug substance and drug product cGMP manufacturing;
|
| • |
AGC Biologics A/S, headquartered in Copenhagen, Denmark, for late-phase clinical and commercial scale drug substance cGMP manufacturing; and
|
| • |
Baxter Oncology GmbH, headquartered in Halle/Westfalen, Germany for late-phase clinical and commercial scale drug product cGMP manufacturing.
|
| • |
nonclinical laboratory tests, animal studies and formulation studies all performed in accordance with the FDA’s good laboratory practices, or GLP, regulations;
|
| • |
submission to the FDA of an IND application for human clinical testing, which must become effective before human clinical trials may begin;
|
| • |
approval by an institutional review board, or IRB, representing each clinical site before each clinical trial may be initiated;
|
| • |
performance of adequate and well-controlled human clinical trials to establish the safety, potency and purity of the product candidate for each proposed indication, in accordance with Good Clinical Practices, or GCP;
|
| • |
preparation and submission to the FDA of a BLA for a biological product requesting marketing for one or more proposed indications, including submission of detailed information on the manufacture and composition of the
product in clinical development and proposed labeling;
|
| • |
review of the product by an FDA advisory committee, if applicable;
|
| • |
one or more FDA inspections of the manufacturing facility or facilities, including those of third parties, at which the product, or components thereof, are produced to assess compliance with current Good Manufacturing
Practices, or cGMP, requirements and to assure that the facilities, methods and controls are adequate to preserve the product’s identity, strength, quality and purity;
|
| • |
FDA audits of the clinical study sites to assure compliance with GCPs, and the integrity of clinical data in support of the BLA;
|
| • |
payment of user fees and securing FDA approval of the BLA and licensure of the new biological product; and
|
| • |
compliance with any post-approval requirements, including the potential requirement to implement a Risk Evaluation and Mitigation Strategy, or REMS, and any post-approval studies required by the FDA.
|
| • |
restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
|
| • |
fines, untitled letters or warning letters or holds on post-approval clinical trials;
|
| • |
refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals;
|
| • |
product seizure or detention, or refusal to permit the import or export of products; or
|
| • |
injunctions or the imposition of civil or criminal penalties.
|
| • |
the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in cash or
kind, in exchange for, or to induce, either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made, in whole or in part, under federal healthcare programs such as the
Medicare and Medicaid programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers, on the one hand, and prescribers, purchasers, formulary managers and other individuals and entities on the other.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the ACA, amended the intent requirement of the federal Anti-Kickback Statute such that a person or entity no longer
needs to have actual knowledge of this statute or specific intent to violate it in order to commit a violation;
|
| • |
the federal civil and criminal false claims, including the civil False Claims Act, or the FCA, and civil monetary penalties laws, which prohibit, among other things, individuals or entities from knowingly presenting,
or causing to be presented, claims for payment from Medicare, Medicaid or other third-party payors that are false or fraudulent, or knowingly making, or causing to be made, a false record or statement material to a false or fraudulent claim to
avoid, decrease, or conceal an obligation to pay money to the federal government. Certain marketing practices, including off-label promotion, also may implicate the FCA. In addition, the ACA codified case law that a claim including items or
services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA.
|
| • |
HIPAA imposes criminal and civil liability, among other things, for executing, or attempting to execute, a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters;
|
| • |
the federal Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health
Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services, or CMS, information related to payments and other transfers of value made to physicians (defined to include doctors, dentists,
optometrists, podiatrists and chiropractors) and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members. Effective January 1, 2022, applicable manufacturers will also be required to
report information regarding payments and transfers of value provided to with physician assistants, nurse practitioners, clinical nurse specialists, anesthesiologist assistants, certified registered nurse anesthetists and certified nurse
midwives during the previous year;
|
| • |
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, and their implementing regulations, which imposes certain obligations, including mandatory contractual terms, with respect
to safeguarding the transmission, security and privacy of individually identifiable health information on covered entities, such as health plans, health care clearinghouses and certain healthcare providers, and their respective business
associates that create, receive, maintain or transmit individually identifiable health information for or on behalf of a covered entity, and their subcontractors that use, disclose, access, or otherwise process individually identifiable
protected health information; and
|
| • |
state and foreign law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers;
state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government that otherwise restrict payments that may
be made to healthcare providers and other potential referral sources; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers, drug pricing
and/or marketing expenditures; state and local laws requiring the registration of pharmaceutical sales representatives; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from
each other in significant ways and often are not preempted by HIPAA, and may not have the same effect, thus complicating compliance efforts.
|
| • |
an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government
healthcare programs;
|
| • |
an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program to 23.1% and 13.0% of the average manufacturer price for most branded and generic drugs, respectively;
|
| • |
a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected;
|
| • |
extension of a manufacturer’s Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations;
|
| • |
expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to certain individuals with income at or below 133% of the federal poverty level, thereby
potentially increasing a manufacturer’s Medicaid rebate liability;
|
| • |
a new Medicare Part D coverage gap discount program, in which manufacturers must now agree to offer 70% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their
coverage gap period, as a condition for a manufacturer’s outpatient drugs to be covered under Medicare Part D;
|
| • |
expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; and
|
| • |
a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research.
|
| C. |
Organizational structure.
|

| D. |
Property, plant and equipment.
|
| Item 5. |
| • |
after reviewing trial results, our collaboration partners may abandon projects that might previously have been believed to be promising;
|
| • |
we, our collaboration partners, or regulators may suspend or terminate clinical trials if the participating subjects or patients are being exposed to unacceptable health risks;
|
| • |
our potential products may not have the desired effects or may include undesirable side effects or other characteristics that preclude regulatory approval or limit their commercial use if approved;
|
| • |
manufacturers may not meet the necessary standards for the production of the product candidates or may not be able to supply the product candidates in a sufficient quantity;
|
| • |
regulatory authorities may find that our clinical trial design or conduct does not meet the applicable approval requirements; and
|
| • |
safety and efficacy results in various human clinical trials reported in scientific and medical literature may not be indicative of results we obtain in our clinical trials.
|
|
Year ended December 31,
|
||||||||||||
|
|
2020
|
2019
|
||||||||||
|
$
|
000
|
£
|
000
|
£
|
000
|
|||||||
|
Revenue
|
41,142
|
30,114
|
25,669
|
|||||||||
|
Research and development expenses
|
(102,204
|
)
|
(74,809
|
)
|
(99,991
|
)
|
||||||
|
Administrative expenses
|
(62,490
|
)
|
(45,740
|
)
|
(44,183
|
)
|
||||||
|
Net other operating income
|
5,795
|
4,242
|
185
|
|||||||||
|
Operating loss
|
(117,757
|
)
|
(86,193
|
)
|
(118,320
|
)
|
||||||
|
Finance income
|
3,017
|
2,208
|
1,510
|
|||||||||
|
Finance costs
|
(4,611
|
)
|
(3,375
|
)
|
(9,379
|
)
|
||||||
|
Non-operating expense
|
(1,594
|
)
|
(1,167
|
)
|
(7,869
|
)
|
||||||
|
Loss before taxes
|
(119,351
|
)
|
(87,360
|
)
|
(126,189
|
)
|
||||||
|
Income tax credit
|
18,125
|
13,267
|
22,258
|
|||||||||
|
Loss for the period
|
(101,226
|
)
|
(74,093
|
)
|
(103,931
|
)
|
||||||
|
Year ended December 31,
|
||||||||||||
|
|
2020
|
2019
|
||||||||||
|
$
|
000
|
£
|
000
|
£
|
000
|
|||||||
|
GSK
|
8,684
|
6,356
|
5,753
|
|||||||||
|
Eli Lilly
|
4,812
|
3,522
|
819
|
|||||||||
|
Genentech
|
27,646
|
20,236
|
19,097
|
|||||||||
|
Total
|
41,142
|
30,114
|
25,669
|
|||||||||
|
Year ended December 31,
|
||||||||||||
|
|
2020
|
2019
|
||||||||||
|
$
|
000
|
£
|
000
|
£
|
000
|
|||||||
|
External research and development expenses:
|
||||||||||||
|
Tebentafusp
|
42,862
|
31,373
|
52,406
|
|||||||||
|
IMC-F106C (PRAME)
|
3,262
|
2,388
|
2,825
|
|||||||||
|
IMC-C103C (MAGE-A4)
|
6,174
|
4,519
|
3,182
|
|||||||||
|
Other programs
|
14,375
|
10,522
|
8,870
|
|||||||||
|
Research expenses
|
663
|
485
|
795
|
|||||||||
|
Total external research and development expenses
|
67,336
|
49,287
|
68,078
|
|||||||||
|
Internal research and development expenses:
|
||||||||||||
|
Headcount related expenses
|
26,694
|
19,539
|
23,320
|
|||||||||
|
Laboratory consumables
|
5,917
|
4,331
|
6,704
|
|||||||||
|
Laboratory equipment expenses
|
2,171
|
1,589
|
1,411
|
|||||||||
|
Other
|
86
|
63
|
478
|
|||||||||
|
Total internal research and development expenses
|
34,868
|
25,522
|
31,913
|
|||||||||
|
Total research and development expenses
|
102,204
|
74,809
|
99,991
|
|||||||||
|
Year ended December 31,
|
||||||||
|
|
2019
|
2018
|
||||||
|
£
|
000
|
£
|
000
|
|||||
|
Revenue
|
25,669
|
23,654
|
||||||
|
Research and development expenses
|
(99,991
|
)
|
(83,575
|
)
|
||||
|
Administrative expenses
|
(44,183
|
)
|
(34,156
|
)
|
||||
|
Other operating income
|
185
|
622
|
||||||
|
Operating loss
|
(118,320
|
)
|
(93,455
|
)
|
||||
|
Other income
|
-
|
4,979
|
||||||
|
Finance income
|
1,510
|
1,140
|
||||||
|
Finance costs
|
(9,379
|
)
|
(842
|
)
|
||||
|
Non-operating (expense)/income
|
(7,869
|
)
|
5,277
|
|||||
|
Loss before taxes
|
(126,189
|
)
|
(88,178
|
)
|
||||
|
Income tax credit
|
22,258
|
16,548
|
||||||
|
Loss for the period
|
(103,931
|
)
|
(71,630
|
)
|
||||
|
Year ended December 31,
|
||||||||
|
|
2019
|
2018
|
||||||
|
£
|
000
|
£
|
000
|
|||||
|
GSK
|
5,753
|
6,079
|
||||||
|
Eli Lilly
|
819
|
8,561
|
||||||
|
Genentech
|
19,097
|
1,461
|
||||||
|
MedImmune
|
-
|
7,553
|
||||||
|
Total
|
25,669
|
23,654
|
||||||
|
Year ended December 31,
|
||||||||
|
|
2019
|
2018
|
||||||
|
£
|
000
|
£
|
000
|
|||||
|
External research and development expenses:
|
||||||||
|
Tebentafusp
|
52,406
|
34,493
|
||||||
|
IMC-F106C (PRAME)
|
2,825
|
1,179
|
||||||
|
IMC-C103C (MAGE-A4)
|
3,182
|
2,315
|
||||||
|
Other programs
|
8,870
|
9,710
|
||||||
|
Research expenses
|
795
|
1,025
|
||||||
|
Total external research and development expenses
|
68,078
|
48,722
|
||||||
|
Internal research and development expenses:
|
||||||||
|
Headcount related expenses
|
23,320
|
23,475
|
||||||
|
Laboratory consumables
|
6,704
|
8,146
|
||||||
|
Laboratory equipment expenses
|
1,411
|
1,589
|
||||||
|
Other
|
478
|
1,643
|
||||||
|
Total internal research and development expenses
|
31,913
|
34,853
|
||||||
|
Total research and development expenses
|
99,991
|
83,575
|
||||||
| B. |
|
Year ended December 31,
|
||||||||||||||||
|
|
2020
|
2019
|
2018
|
|||||||||||||
|
$
|
000
|
£
|
000
|
£
|
000
|
£
|
000
|
|||||||||
|
Cash and cash equivalents at beginning of the year
|
101,052
|
73,966
|
124,385
|
82,883
|
||||||||||||
|
Net cash flows used in operating activities
|
(82,756
|
)
|
(60,574
|
)
|
(101,376
|
)
|
(16,626
|
)
|
||||||||
|
Net cash flows from / (used in) investing activities
|
638
|
467
|
(4,137
|
)
|
58,014
|
|||||||||||
|
Net cash flows from financing activities
|
158,399
|
115,941
|
55,127
|
101
|
||||||||||||
|
Net foreign exchange difference on cash held
|
(115
|
)
|
(84
|
)
|
(33
|
)
|
13
|
|||||||||
|
Cash and cash equivalents at end of the year
|
177,218
|
129,716
|
73,966
|
124,385
|
||||||||||||
| • |
continue to advance the development of our clinical trials and pre-clinical programs;
|
| • |
continue to invest in our soluble TCR platforms to conduct research to identify novel technologies;
|
|
•
|
change or add additional suppliers;
|
| • |
add additional infrastructure to our quality control, quality assurance, legal, compliance and other groups to support our operations as we progress product candidates toward commercialization;
|
|
•
|
seek to attract and retain skilled personnel;
|
|
•
|
create additional infrastructure to support our operations as a public company listed in the United States and our product development and planned future commercialization efforts;
|
|
•
|
seek marketing approvals and reimbursement for our product candidates;
|
|
•
|
establish a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain marketing approval;
|
|
•
|
seek to identify and validate additional product candidates;
|
|
•
|
acquire or in-license other product candidates and technologies;
|
|
•
|
maintain, protect, defend, enforce and expand our intellectual property portfolio; and
|
|
•
|
experience any delays, interruptions or encounter issues with any of the above, including any delays or other impacts as a result of the COVID-19 pandemic.
|
|
•
|
the progress, timing, scope and costs of our clinical trials, including the ability to timely initiate clinical sites, enroll subjects and manufacture soluble bispecific TCR product candidates for our ongoing,
planned and potential future clinical trials;
|
|
•
|
the time and costs required to perform research and development to identify and characterize new product candidates from our research programs;
|
|
•
|
the time and cost necessary to obtain regulatory authorizations and approvals that may be required by regulatory authorities to execute clinical trials or commercialize our products;
|
|
•
|
our ability to successfully commercialize our product candidates, if approved;
|
|
•
|
our ability to have clinical and commercial products successfully manufactured consistent with FDA, EMA and other authorities’ regulations;
|
|
•
|
the amount of sales and other revenues from product candidates that we may commercialize, if any, including the selling prices for such potential products and the availability of adequate third-party coverage and
reimbursement for patients;
|
|
•
|
the sales and marketing costs associated with commercializing our products, if approved, including the cost and timing of building our marketing and sales capabilities;
|
|
•
|
the cost of building, staffing and validating our manufacturing processes, which may include capital expenditure;
|
|
•
|
the terms and timing of any revenue from our existing collaborations;
|
|
•
|
the costs of operating as a public company;
|
|
•
|
the time and cost necessary to respond to technological, regulatory, political and market developments;
|
|
•
|
the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights;
|
|
•
|
the costs, associated with, and terms and timing of, any future any potential acquisitions, strategic collaborations, licensing agreements or other arrangements that we may establish; and
|
|
•
|
the inability of clinical sites to enroll patients as healthcare capacities are required to cope with natural disasters, epidemics or other health system emergencies, such as the COVID-19 pandemic.
|
|
•
|
whether achievement of a development milestone is highly susceptible to factors outside the entity’s influence, such as milestones involving the judgment or actions of third parties, including regulatory bodies or
the customer;
|
|
•
|
whether the uncertainty about the achievement of the milestone is not expected to be resolved for a long period of time;
|
|
•
|
whether we can reasonably predict that a milestone will be achieved based on previous experience; and.
|
|
•
|
the complexity and inherent uncertainty underlying the achievement of the milestone.
|
|
•
|
adjustments arising from a change in the estimate of when the performance obligation will have been completed.
|
|
•
|
adjustment to revenue that affects deferred revenue;
|
|
•
|
a change in the estimate of the transaction price due to changes in the assessment of whether variable consideration is constrained because it is not considered probable of being received; and
|
|
•
|
the recognition of revenue.
|
|
•
|
past history and experience with similar contracts.
|
|
•
|
unexpected fluctuations in planned spend.
|
|
•
|
changes to project timelines.
|
|
•
|
the data generated from our research and development programs;
|
|
•
|
our future operating performance, prospects and business strategy;
|
|
•
|
the material risks related to our business and industry;
|
|
•
|
the lack of an active public market for our ordinary and convertible preferred shares;
|
|
•
|
the market performance of publicly traded companies in the life science and biotechnology sectors;
|
|
•
|
the prices at which we issued ordinary and preferred shares and the superior rights and preferences of the preferred shares relative to the ordinary shares at the time of each grant; and
|
|
•
|
the likelihood of achieving a liquidity events for the holders of our ordinary shares, series A preferred and series B preferred shares and G shares, such as an initial public offering, given prevailing market
conditions.
|
| C. |
Research and Development
|
| D. |
Trend Information
|
| E. |
Off-Balance Sheet Arrangements
|
| F. |
Tabular Disclosure of Contractual Obligations
|
|
As at December 31, 2020
|
Less than 1
year
|
1-3 years
|
3-5 years
|
More than
5 years
|
Total
|
|||||||||||||||
|
Lease liabilities – existing
|
3,529
|
5,322
|
4,286
|
32,600
|
45,737
|
|||||||||||||||
|
Lease liabilities – contingent
|
-
|
2,254
|
2,471
|
1,841
|
6,566
|
|||||||||||||||
|
Manufacturing
|
2,824
|
500
|
-
|
-
|
3,324
|
|||||||||||||||
|
Capital Commitments
|
77
|
-
|
-
|
-
|
77
|
|||||||||||||||
|
Total contractual obligations (in thousands, pounds)
|
6,430
|
8,076
|
6,757
|
34,441
|
55,704
|
|||||||||||||||
|
Total contractual obligations (in thousands, U.S. Dollars)
|
8,785
|
11,033
|
9,231
|
47,053
|
76,103
|
|||||||||||||||
| G. |
Safe Harbor
|
| Item 6. |
| A. |
|
Name
|
|
Age
|
|
Position(s)
|
|
Executive Officers:
|
|
|
||
|
Bahija Jallal, Ph.D.
|
|
59
|
|
Chief Executive Officer and Director
|
|
Brian Di Donato
|
|
54
|
|
Chief Financial Officer and Head of Strategy
|
|
David Berman, M.D., Ph.D.
|
|
50
|
|
Head of Research and Development
|
|
Non-Executive Directors:
|
|
|
||
|
Professor Sir John Bell
|
|
68
|
|
Chairman of the Board of Directors
|
|
Travis Coy
|
|
40
|
|
Director
|
|
Roy S. Herbst, M.D., Ph.D
|
|
58
|
|
Director
|
|
Robert Perez
|
|
56
|
|
Director
|
|
Kristine Peterson
|
|
61
|
|
Director
|
|
Professor Sir Peter Ratcliffe
|
|
66
|
|
Director
|
|
B.
|
Compensation
|
| 1. |
Annual Board of Directors Service Retainer:
|
|
a.
|
All Eligible Directors: $40,000
|
|
b.
|
Independent Chair of the Board of Directors Service Retainer (in addition to Eligible Director Service Retainer): $30,000
|
|
2.
|
Annual Committee Member Service Retainer:
|
|
a.
|
Member of the Audit Committee: $7,500
|
|
b.
|
Member of the Remuneration Committee: $5,000
|
|
c.
|
Member of the Nominating and Corporate Governance Committee: $4,000
|
|
3.
|
Annual Committee Chair Service Retainer (in addition to Annual Committee Member Service Retainer):
|
|
a.
|
Chair of the Audit Committee: $7,500
|
|
b.
|
Chair of the Remuneration Committee: $5,000
|
|
c.
|
Chair of the Nominating and Corporate Governance Committee: $4,000
|
|
Name
|
Ordinary Share Underlying Option
|
Exercise Price
|
Grant
Date
|
Expiration
Date
|
||||||||||||
|
Executive Director
|
||||||||||||||||
|
Bahija Jallal, Ph.D.
|
5,669
|
$
|
17.4643
|
October 30, 2020
|
October 29, 2030
|
|||||||||||
|
Non-Executive Directors
|
||||||||||||||||
|
Professor Sir John Bell
|
18,215
|
$
|
17.4643
|
November 16, 2020
|
November 15, 2030
|
|||||||||||
|
Travis Coy
|
-
|
-
|
-
|
-
|
||||||||||||
|
Roy Herbst, M.D., Ph.D.
|
-
|
-
|
-
|
-
|
||||||||||||
|
Robert Perez
|
-
|
-
|
-
|
-
|
||||||||||||
|
Kristine Peterson
|
11,520
|
$
|
17.4643
|
November 16, 2020
|
November 15, 2030
|
|||||||||||
|
Professor Sir Peter Ratcliffe
|
-
|
-
|
-
|
-
|
||||||||||||
| • |
an attempt to transfer, assign or encumber the option (save for a transfer to a personal representative on death);
|
| • |
a performance condition failing to be met that results in the entire option being incapable of exercise;
|
| • |
the date stated in the relevant option certificate;
|
| • |
the first anniversary of an option holder’s death;
|
| • |
90 days after the option holder ceases to be employed by the company;
|
| • |
if the board of directors uses its discretion to permit early exercise of an option within a defined period determined by the board of directors, the expiry of such period;
|
| • |
40 days after the completion of a Takeover or an Asset Sale (both as defined below) (or immediately after completion if option holders are given the opportunity to exercise their options by the board of directors
prior to completion);
|
| • |
40 days after a reorganization of the company if a replacement option is offered in the acquirer as part of the reorganization; or
|
|
•
|
the option holder becoming bankrupt.
|
| • |
they are a Good Leaver, in which case they may exercise their vested option and 50% of their unvested option (calculated as at the date the option holder ceased to employed) for a period ending 90 days after becoming
a Leaver, or 12 months from the date of death if the reason for leaving is due to an option holder’s death; or
|
| • |
they are Bad Leaver, in which case they may exercise their vested option (calculated as at the date the option holder ceased to employed) for a period ending 90 days after becoming a Leaver; or
|
| • |
the board of directors determines otherwise.
|
| • |
an attempt to transfer, assign or encumber the option (save for a transfer to a personal representative on death);
|
| • |
a performance condition failing to be met that results in the entire option being incapable of exercise;
|
| • |
the date stated in the relevant option certificate;
|
| • |
the first anniversary of an option holder’s death;
|
| • |
90 days after the option holder ceases to be employed or engaged by the company;
|
| • |
if the board of directors uses its discretion to permit early exercise of an option within a defined period determined by the board of directors, the expiry of such period;
|
| • |
40 days after the completion of a Takeover or an Asset Sale (or immediately after completion if option holders are given the opportunity to exercise their options by the board of directors prior to completion); or
|
| • |
the option holder becoming bankrupt.
|
| • |
they are a Good Leaver, in which case they may exercise their vested option and 50% of their unvested option (calculated as at the date the option holder ceased to employed) for a period ending 90 days after
becoming a Leaver, or 12 months from the date of death if the reason for leaving is due to an option holder’s death; or
|
| • |
they are Bad Leaver, in which case they may exercise their vested option (calculated as at the date the option holder ceased to employed) for a period ending 90 days after becoming a Leaver; or
|
| • |
the board of directors determines otherwise.
|
|
C.
|
Board Practices
|
| • |
Class I consists of Travis Coy and Peter Ratcliffe, whose terms will expire at our first annual general meeting held in 2022;
|
| • |
Class II consists of Robert Perez and Kris Peterson, whose terms will expire at our second annual general meeting held in 2023; and
|
| • |
Class III consists of John Bell and Bahija Jallal, whose terms will expire at our third annual general meeting held in 2024.
|
| • |
reviewing, modifying and overseeing the company’s overall compensation strategy and policies;
|
| • |
reviewing and approving the compensation and other terms of employment of our Chief Executive Officer;
|
| • |
reviewing and approving all elements of the compensation and other terms of employment of the executive officers and other senior management reporting directly to the Chief Executive Officer;
|
| • |
reviewing and recommending to the board of directors for its approval the type and amount of compensation to be paid or awarded to members of the board of directors;
|
| • |
undertaking sole responsibility for the appointment, authority to select, retain, and terminate any compensation and oversight of the work of compensation consultants, legal counsel, or any other advisors engaged for
the purpose of advising the remuneration committee;
|
| • |
exercising full power and authority to adopt, amend, terminate, and administer our equity award, pension, and profit sharing plans, incentive plans, bonus plans, executive benefit plans, stock purchase plans, deferred
compensation plans and other similar programs;
|
| • |
when required, reviewing and discussing with management our Compensation Discussion and Analysis section of our annual reports, registration statements, proxy statements, or information statements filed with the SEC;
|
| • |
reviewing and discussing with management any conflicts of interest raised; and
|
| • |
overseeing the preparation of any report required by applicable U.S. and U.K. rules and regulations to be included in our public filings relating to compensation policy and practices, including but not limited to the
directors’ remuneration report required under the Companies Act.
|
| • |
identifying and evaluating candidates, including nomination of incumbent directors for re-election and nominees recommended by shareholders to serve on the board of directors;
|
| • |
making recommendations to the board of directors regarding nominees for directors at the next annual general meeting;
|
| • |
periodically reviewing the performance of the board of directors, including committees of the board of directors and management;
|
| • |
overseeing the board of directors' committee structure and operations, including authority to delegate to subcommittees and committee reporting to the board of directors;
|
| • |
reviewing with the Chief Executive Officer the succession plans for our executive officers;
|
| • |
instituting plans or programs for the continuing education of directors and orientation of new directors, as it deems appropriate; and
|
| • |
periodically reviewing the processes and procedures to provide information to the board of directors and its committees.
|
| D. |
Employees
|
|
|
At December 31,
|
|||||||||||
|
|
2018
|
2019
|
2020(1)
|
|||||||||
|
Function:
|
||||||||||||
|
Administrative
|
67
|
67
|
55
|
|||||||||
|
Research and development
|
394
|
392
|
236
|
|||||||||
|
Total
|
461
|
459
|
291
|
|||||||||
|
Geography:
|
||||||||||||
|
United Kingdom
|
422
|
409
|
242
|
|||||||||
|
European Union
|
2
|
3
|
2
|
|||||||||
|
United States
|
37
|
47
|
47
|
|||||||||
|
Total
|
461
|
459
|
291
|
|||||||||
|
(1)
|
As a result of the corporate restructuring, which was completed in the second quarter of 2020, our overall headcount was reduced by 78.
|
|
E.
|
Share Ownership
|
| Item 7. |
| A. |
Major Shareholders
|
| • |
each beneficial owner of 5% or more of our outstanding ordinary shares and non-voting ordinary shares;
|
| • |
each of our directors and executive officers; and
|
| • |
all of our directors and executive officers as a group.
|
|
Name of Beneficial Owner
|
Number of
Ordinary Shares
Beneficially
Owned (#)
|
Percent of
Ordinary
Shares
Beneficially
Owned (%)
|
||||||
|
5% or Greater Shareholders:
|
||||||||
|
Entities affiliated with General Atlantic(1)
|
4,922,575
|
11.2
|
%
|
|||||
|
Entities affiliated with Baker Brothers(2)
|
3,352,357
|
7.7
|
%
|
|||||
|
Eli Lilly S.A.(3)
|
2,548,145
|
5.8
|
%
|
|||||
|
Entities affiliated with Fidelity(4)
|
2,511,530
|
5.7
|
%
|
|||||
|
Nicholas John Cross(5)
|
2,369,610
|
5.4
|
%
|
|||||
|
Ian Laing(6)
|
2,364,420
|
5.4
|
%
|
|||||
|
Malin Life Sciences Holdings Limited(7)
|
2,359,425
|
5.4
|
%
|
|||||
|
Executive Officers and Directors:
|
||||||||
|
Bahija Jallal, Ph.D.(8)
|
519,867
|
1.2
|
%
|
|||||
|
Brian Di Donato(9)
|
19,230
|
*
|
||||||
|
David Berman, M.D., Ph.D.(10)
|
519,865
|
1.2
|
% | |||||
|
Professor Sir John Bell(11)
|
75,237
|
*
|
||||||
|
Travis Coy
|
—
|
—
|
||||||
|
Roy Herbst(12)
|
10,620
|
*
|
||||||
|
Robert Perez
|
—
|
—
|
||||||
|
Kristine Peterson(13)
|
25,298
|
*
|
||||||
|
All current directors and executive officers as a group
(8 persons)(14)
|
1,170,117
|
2.7
|
%
|
|||||
| * |
Represents beneficial ownership of less than one percent.
|
| (1) |
Consists of 4,922,575 ADSs held by GA IMC Holding, L.P. The limited partners that share beneficial ownership of the shares held by GA IMC Holding are the following
General Atlantic investment funds: General Atlantic Partners (Bermuda) EU, L.P. (“GAP EU”), General Atlantic Partners (Bermuda) IV, L.P. (“GAP IV”) , GAP Coinvestments III, LLC (“GAPCO III”), GAP Coinvestments IV, LLC (“GAPCO IV”), GAP
Coinvestments V, LLC (“GAPCO V”) and GAP Coinvestments CDA, LLC (“GAPCO CDA”). The general partner of GAP EU and GAP IV is General Atlantic GenPar (Bermuda), L.P. (“GenPar Bermuda”). GAP (Bermuda) Limited (“GAP (Bermuda) Limited”) is the
general partner of GenPar Bermuda. General Atlantic’s address is c/o Conyers Client Services (Bermuda) Limited, Clarendon House, 2 Church Street, Hamilton MM II,
Bermuda.
|
| (2) |
The information shown is based, in part, upon disclosures filed on a Schedule 13G on February 16, 2020 filed jointly by Baker Bros. Advisors LP (the “Adviser”), Baker Bros. Advisors (GP) LLC (the “Adviser GP”),
Felix J. Baker and Julian C. Baker (collectively, the “Reporting Persons”). The number reported consists of 2,520,730 ADSs and 831,627 ordinary shares issuable upon conversion of 831,627 non-voting ordinary shares directly held by the funds.
3,104,143 ADSs are held by Baker Brothers Life Sciences, L.P. and 248,214 ADSs are held by 667,L.P.The address of Baker Bros. Advisors LP is 860 Washington Street, 3rd
Floor, New York, NY 10014, United States.
|
| (3) |
Consists of 2,548,145 ADSs held by Eli Lilly S.A. Eli Lilly S.A.’s address is 16, Chemin des Coquelicots, 12 Geneva, Switzerland.
|
| (4) |
Consists of 2,511,530 ADSs held by Fidelity Select Portfolios: Select Biotechnology Portfolio, Fidelity Mt. Vernon Street Trust: Fidelity Growth Company Fund, Fidelity Growth Company Commingled Pool, Fidelity Mt.
Vernon Street Trust: Fidelity Series Growth Company Fund, Fidelity Advisor Series VII: Fidelity Advisor Biotechnology Fund, Fidelity Securities Fund: Fidelity Blue Chip Growth Fund, Fidelity Securities Fund: Fidelity Series Blue Chip Growth
Fund, FIAM Target Date Blue Chip Growth Commingled Pool and Fidelity Blue Chip Growth Commingled Pool.
|
| (5) |
Consists of 2,369,610 ADSs held by Mr. Cross.
|
| (6) |
Consists of (a) 1,914,102 ADSs held by Mr. Laing, (b) options to purchase 1,038 ordinary shares that are or will be immediately exercisable within 60 days of February 9, 2021 held by Mr. Laing, and
(c) 449,280 ADS’s held by Mr. Laing’s spouse.
|
| (7) |
Consists of 2,359,425 ADSs held by Malin Life Sciences Holdings Limited. Malin Life Sciences Holdings Limited’s address is The Lennox Building, 50 Richmond Street South, Dublin D02 FK02, Ireland.
|
| (8) |
Consists of 519,867 ordinary shares underlying options that are exercisable within 60 days of February 9, 2021 held by Dr. Jallal.
|
|
(9)
|
Consists of 19,230 ADSs held by Mr. Di Donato.
|
| (10) |
Consists of 519,865 ordinary shares underlying options that are exercisable within 60 days of February 9, 2021 held by Dr. Berman.
|
| (11) |
Consists of (a) 13,452 ADSs and (b) options to purchase 61,785 ordinary shares that are or will be immediately exercisable within 60 days of February 9, 2021 held by Professor Sir John Bell.
|
| (12) |
Consists of 10,620 ordinary shares underlying options that are exercisable within 60 days of February 9, 2021 held by Mr. Herbst.
|
| (13) |
Consists of 25,298 ordinary shares underlying options that are exercisable within 60 days of February 9, 2021 held by Ms. Peterson.
|
| (14) |
Consists of (a) 32,682 ADSs and (b) options to purchase 1,137,435 ordinary shares that are or will be immediately exercisable within 60 days of February 9, 2021.
|
|
B.
|
Related Party Transactions
|
|
Purchaser
|
Number
of ADSs
|
|||
|
Entities affiliated with General Atlantic
|
950,000
|
|||
|
Entities affiliated with Baker Brothers
|
1,689,102
|
|||
|
Entities affiliated with Fidelity
|
1,350,000
|
|||
|
Participants
|
Series C Preferred
Shares (#)
|
Ordinary Shares (#)
|
||||||
|
Entities affiliated with Baker Brothers
|
274,574
|
4,965
|
||||||
|
Entities affiliated with General Atlantic
|
219,659
|
18,963
|
||||||
|
Eli Lilly S.A.
|
—
|
23,238
|
||||||
|
Participants
|
Series B
Preferred
Shares (#)
|
|||
|
Entities affiliated with General Atlantic(1)
|
555,893
|
|||
|
Eli Lilly S.A.
|
71,588
|
|||
|
(1)
|
These shares were purchased by GA IMC Holding, L.P.
|
| C. |
Interests of Experts and Counsel
|
| Item 8. |
Financial Information.
|
| A. |
Consolidated Statements and Other Financial Information
|
| B. |
Significant Changes
|
| Item 9. |
The Offer and Listing.
|
| A. |
Offer and Listing Details
|
| B. |
Plan of Distribution
|
| C. |
Markets
|
| D. |
Selling Shareholders
|
| E. |
Dilution
|
| F. |
Expenses of the Issue
|
| Item 10. |
Additional Information.
|
| A. |
| B. |
| C. |
| D. |
| E. |
| • |
banks, insurance companies, and certain other financial institutions;
|
| • |
U.S. expatriates and certain former citizens or long-term residents of the United States;
|
| • |
dealers or traders in securities who use a mark-to-market method of tax accounting;
|
| • |
persons holding ordinary shares or ADSs as part of a hedging transaction, “straddle,” wash sale, conversion transaction or integrated transaction or persons entering into a constructive sale with respect to ordinary shares or ADSs;
|
| • |
persons whose “functional currency” for U.S. federal income tax purposes is not the U.S. dollar;
|
| • |
brokers, dealers or traders in securities, commodities or currencies;
|
| • |
tax-exempt entities or government organizations;
|
| • |
S corporations, partnerships, or other entities or arrangements classified as partnerships for U.S. federal income tax purposes (and investors therein);
|
| • |
regulated investment companies or real estate investment trusts;
|
| • |
persons who acquired our ordinary shares or ADSs pursuant to the exercise of any employee stock option or otherwise as compensation;
|
| • |
persons holding shares or ADSs in connection with a trade or business outside the United States;
|
| • |
persons that own or are deemed to own ten percent or more of our shares (by vote or value); and
|
| • |
persons holding our ordinary shares or ADSs in connection with a trade or business, permanent establishment, or fixed base outside the United States.
|
| • |
the excess distribution or gain will be allocated ratably over a U.S. Holder’s holding period for the ordinary shares or ADSs;
|
| • |
the amount allocated to the taxable year of the disposition or distribution (as applicable), and any taxable year prior to the first taxable year in which we became a PFIC, will be treated as ordinary income; and
|
| • |
the amount allocated to each other year will be subject to the highest tax rate in effect for that year and the interest charge generally applicable to underpayments of tax will be imposed on the resulting tax attributable to each such
year.
|
| • |
persons who are connected with the company;
|
| • |
financial institutions;
|
| • |
insurance companies;
|
| • |
charities or tax-exempt organizations;
|
| • |
collective investment schemes;
|
| • |
pension schemes;
|
| • |
market makers, intermediaries, brokers or dealers in securities;
|
| • |
persons who have (or are deemed to have) acquired their ADSs by virtue of an office or employment or who are or have been officers or employees of the company or any of its affiliates; and
|
| • |
individuals who are subject to U.K. taxation on a remittance basis.
|
| F. |
| G. |
| H. |
| I. |
| Item 11. |
Quantitative and Qualitative Disclosures About Market Risk.
|
| Item 12. |
Description of Securities Other than Equity Securities.
|
| A. |
| B. |
| C. |
| D. |
|
Service
|
|
Fee
|
|
Issuance of ADSs (e.g., an issuance of ADS upon a deposit of ordinary shares or upon a change in the ADS(s)-to-ordinary shares ratio, or for any other reason), excluding
ADS issuances as a result of distributions of ordinary shares
|
|
Up to $0.05 per ADS issued
|
|
|
||
|
Cancellation of ADSs (e.g., a cancellation of ADSs for delivery of deposited property or upon a change in the ADS(s)-to-ordinary shares ratio, or for any other reason)
|
|
Up to $0.05 per ADS cancelled
|
|
|
||
|
Distribution of cash dividends or other cash distributions (e.g., upon a sale of rights and other entitlements)
|
|
Up to $0.05 per ADS held
|
|
|
||
|
Distribution of ADSs pursuant to (i) share dividends or other distributions, or (ii) exercise of rights to purchase additional ADSs
|
|
Up to $0.05 per ADS held
|
|
|
||
|
Distribution of securities other than ADSs or rights to purchase additional ADSs (e.g., upon a spin-off)
|
|
Up to $0.05 per ADS held
|
|
|
||
|
ADS services
|
|
Up to $0.05 per ADS held on the applicable record date(s) established by the depositary
|
| • |
taxes (including applicable interest and penalties) and other governmental charges;
|
| • |
the registration fees as may from time to time be in effect for the registration of ordinary shares on the share register and applicable to transfers of ordinary shares to or from the name of the custodian, the depositary or any nominees
upon the making of deposits and withdrawals, respectively;
|
| • |
certain cable, telex and facsimile transmission and delivery expenses;
|
| • |
the expenses and charges incurred by the depositary in the conversion of foreign currency;
|
| • |
the fees and expenses incurred by the depositary in connection with compliance with exchange control regulations and other regulatory requirements applicable to ordinary shares, ADSs and ADRs; and
|
| • |
the fees and expenses incurred by the depositary, the custodian, or any nominee in connection with the servicing or delivery of deposited property.
|
| Item 13. |
Defaults, Dividend Arrearages and Delinquencies.
|
| Item 14. |
Material Modifications to the Rights of Security Holders and Use of Proceeds.
|
| A. |
Not applicable.
|
| B. |
.Not applicable.
|
| C. |
Not applicable.
|
| D. |
Not applicable.
|
| E. |
Use of Proceeds.
|
| Item 15. |
Disclosure Controls and Procedures.
|
| B. |
| C. |
| D. |
| Item 16. |
[Reserved]
|
| Item 16A. |
Audit Committee Financial Expert.
|
| Item 16B. |
Code of Business Conduct and Ethics.
|
|
|
Year Ended December 31, | |||||||
|
2020
|
2019
|
|||||||
| £ | 000 | £ | 000 | |||||
|
Audit fees
|
£
|
470
|
£
|
183
|
||||
|
Audit-related fees
|
237
|
—
|
||||||
|
Total
|
£
|
707
|
£
|
183
|
||||
| Item 16D. |
Exemptions from the Listing Standards for Audit Committees.
|
| Item 16E. |
Purchases of Equity Securities by the Issuer and Affiliated Purchasers.
|
| Item 16F. |
Change in Registrant’s Certifying Accountant.
|
| Item 16G. |
Corporate Governance.
|
| Item 16H. |
Mine Safety Disclosure.
|
| Item 17. |
Financial Statements.
|
| Item 18. |
Financial Statements.
|
Not applicable.
|
Exhibit
|
Description
|
Incorporation by Reference
|
||||||||
|
Schedule/
Form
|
File
Number
|
Exhibit
|
File Date
|
|||||||
|
Articles of Association of Immunocore Holdings plc.
|
||||||||||
|
Deposit Agreement.
|
||||||||||
|
2.3*
|
Form of American Depositary Receipt (included in Exhibit 2.2).
|
|||||||||
|
Description of Securities.
|
||||||||||
|
Subscription Agreement between the Registrant and the Bill & Melinda Gates Foundation, dated February 3, 2021.
|
F-1/A
|
333-252166
|
4.3
|
02/03/21
|
||||||
|
Research Collaboration and License Agreement, dated as of June 14, 2013, by and among the Registrant, Genentech, Inc. and F. Hoffman-La Roche Ltd, as amended on September 27, 2016.
|
F-1
|
333-252166
|
10.5
|
01/15/21
|
||||||
|
Collaboration and License Agreement, dated as of June 29, 2013, between the Registrant and GlaxoSmithKline Intellectual Property Development Ltd.
|
F-1
|
333-252166
|
10.6
|
01/15/21
|
||||||
|
Development and License Agreement, dated as of July 11, 2014, between the Registrant and Eli Lilly and Company, as amended on December 21, 2016, September 20, 2017 and December 19, 2018.
|
F-1
|
333-252166
|
10.7
|
01/15/21
|
||||||
|
License Agreement, dated as of September 27, 2016, between the Registrant and Genentech, Inc.
|
F-1
|
333-252166
|
10.8
|
01/15/21
|
||||||
|
License and Collaboration Agreement, dated as of November 15, 2018, by and among the Registrant, Genentech, Inc. and F. Hoffman-La Roche Ltd.
|
F-1
|
333-252166
|
10.9
|
01/15/21
|
||||||
|
Convertible Loan Note Purchase Agreement, dated as of September 13, 2017, between the Registrant and the Bill & Melinda Gates Foundation.
|
F-1
|
333-252166
|
10.10
|
01/15/21
|
||||||
|
Amended and Restated Global Access Commitments Agreement, dated as of March 2, 2020, between the Registrant and the Bill & Melinda Gates Foundation.
|
F-1
|
333-252166
|
10.11
|
01/15/21
|
||||||
|
First Amendment to the Amended and Restated Global Access Commitments Agreement, dated
as of February 3, 2021, between the Registrant and the Bill & Melinda Gates Foundation.
|
F-1/A
|
333-252166
|
10.12
|
02/03/21
|
||||||
|
Lease, dated as of March 28, 2017, between the Registrant and MEPC MILTON PARK NO. 1 LIMITED and MEPC MILTON PARK NO. 2 LIMITED, on behalf of MEPC Milton LP.
|
F-1
|
333-252166
|
10.13
|
01/15/21
|
||||||
|
Lease, dated as of December 28, 2017, between the Registrant and MEPC MILTON PARK NO. 1 LIMITED and MEPC MILTON PARK NO. 2 LIMITED, on behalf of MEPC Milton LP.
|
F-1
|
333-252166
|
10.14
|
01/15/21
|
||||||
|
Lease, dated as of March 28, 2017, between the Registrant and MEPC MILTON PARK NO. 1 LIMITED and MEPC MILTON PARK NO. 2 LIMITED, on behalf of MEPC Milton LP.
|
F-1
|
333-252166
|
10.15
|
01/15/21
|
||||||
|
Assignment and Exclusive License, dated as of January 28, 2015, between the Registrant and Adaptimmune Limited.
|
F-1
|
333-252166
|
10.16
|
01/15/21
|
|
Loan and Security Agreement, dated as of November 6, 2020, among the Registrant, Oxford
Finance Luxembourg S.ˆ r.l., and the lenders listed on Schedule 1.1 thereof.
|
F-1
|
333-252166
|
10.17
|
01/15/21
|
||||||
|
Employment Agreement between the Registrant and Bahija Jallal, Ph.D., dated January 29, 2021.
|
F-1/A
|
333-252166
|
10.18
|
02/01/21
|
||||||
|
Deed of Termination of Convertible Loan Note Purchase Agreement, dated as of February 3, 2021, between the Registrant and the Bill & Melinda Gates Foundation.
|
F-1/A
|
333-252166
|
10.19
|
02/03/21
|
||||||
|
Form of Deed of Indemnity between the Registrant and each of its directors.
|
F-1
|
333-252166
|
10.1
|
01/15/21
|
||||||
|
Form of Deed of Indemnity between the Registrant and each of its executive officers.
|
F-1
|
333-252166
|
10.2
|
01/15/21
|
||||||
|
Immunocore Holdings plc 2021 Equity Incentive Plan. and
Non-Employee Sub Plan to the Immunocore Holdings plc 2021 Equity Incentive Plan
|
||||||||||
|
Subsidiaries of the Registrant.
|
||||||||||
|
Certification by the Principal Executive Officer pursuant to Securities Exchange Act Rules 13a-14(a) and 15d-14(a) as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
|
||||||||||
|
Certification by the Principal Financial Officer pursuant to Securities Exchange Act Rules 13a-14(a) and 15d-14(a) as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
|
||||||||||
|
Certification by the Principal Executive Officer and the Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
|
|
101.INS*
|
XBRL Instance Document
|
||||||||
|
101.SCH*
|
XBRL Taxonomy Extension Schema Document
|
|||||||||
|
101.CAL*
|
XBRL Taxonomy Extension Calculation Linkbase Document
|
|||||||||
|
101.DEF*
|
XBRL Taxonomy Extension Definition Linkbase Document
|
|||||||||
|
101.LAB*
|
XBRL Taxonomy Extension Label Linkbase Document
|
|||||||||
|
101.PRE*
|
XBRL Taxonomy Extension Presentation Linkbase Document
|
| * |
Filed herewith.
|
| ** |
Furnished herewith.
|
| † |
Certain portions of this exhibit (indicated by asterisks) have been redacted in accordance with Regulation S-K, Item 601(b)(10).
|
| # |
Indicates a management contract or any compensatory plan, contract or arrangement.
|
|
IMMUNOCORE HOLDINGS PLC
|
||
|
By:
|
/s/ Bahija Jallal
|
|
|
Bahija Jallal, Ph.D.
|
||
|
Chief Executive Officer
(Principal Executive Officer)
|
||
| Date: March 25, 2021 |
|
Page
|
|
|
F‑2
|
|
|
F‑3
|
|
|
F‑4
|
|
|
F‑5
|
|
|
F‑6
|
|
|
F‑7
|
|
2020
|
2019
|
2018
|
||||||||||||||
|
Notes
|
£
|
’000
|
£
|
’000
|
£
|
’000
|
||||||||||
|
Revenue
|
2
|
30,114
|
25,669
|
23,654
|
||||||||||||
|
Total revenue
|
30,114
|
25,669
|
23,654
|
|||||||||||||
|
Net other operating income
|
5
|
4,242
|
185
|
622
|
||||||||||||
|
Research and development costs
|
3
|
(74,809
|
)
|
(99,991
|
)
|
(83,575
|
)
|
|||||||||
|
Administrative expenses
|
3
|
(45,740
|
)
|
(44,183
|
)
|
(34,156
|
)
|
|||||||||
|
Operating loss
|
(86,193
|
)
|
(118,320
|
)
|
(93,455
|
)
|
||||||||||
|
Other income
|
—
|
—
|
4,979
|
|||||||||||||
|
Finance income
|
6
|
2,208
|
1,510
|
1,140
|
||||||||||||
|
Finance costs
|
7
|
(3,375
|
)
|
(9,379
|
)
|
(842
|
)
|
|||||||||
|
Non-operating (expense) / income
|
(1,167
|
)
|
(7,869
|
)
|
5,277
|
|||||||||||
|
Loss before taxation
|
(87,360
|
)
|
(126,189
|
)
|
(88,178
|
)
|
||||||||||
|
Income tax credit
|
8
|
13,267
|
22,258
|
16,548
|
||||||||||||
|
Loss for the year
|
(74,093
|
)
|
(103,931
|
)
|
(71,630
|
)
|
||||||||||
|
Other comprehensive (expense) / income
|
||||||||||||||||
|
Other comprehensive (expense) / income that are or may be reclassified to profit or loss in subsequent periods (net of tax):
|
||||||||||||||||
|
Exchange differences on translation of foreign operations
|
195
|
(99
|
)
|
72
|
||||||||||||
|
Income tax effect relating to the components of other comprehensive income
|
8
|
—
|
—
|
3,634
|
||||||||||||
|
Total other comprehensive (expense) / income for the year, net of tax
|
195
|
(99
|
)
|
3,706
|
||||||||||||
|
Total comprehensive loss for the year, net of tax
|
(73,898
|
)
|
(104,030
|
)
|
(67,924
|
)
|
||||||||||
|
Basic and diluted loss per share - £
|
9
|
(2.79
|
)
|
(4.66
|
)
|
(3.32
|
)
|
|||||||||
|
Notes
|
2020
£’000
|
2019
£’000
|
||||||||||
|
Non-current assets
|
||||||||||||
|
Property, plant and equipment
|
11
|
13,754
|
18,302
|
|||||||||
|
Right of use assets
|
12
|
23,093
|
36,578
|
|||||||||
|
Investment in sub-lease
|
12
|
776
|
591
|
|||||||||
|
Other non-current financial assets
|
13
|
4,410
|
4,390
|
|||||||||
|
Deferred tax asset
|
8
|
2,230
|
1,507
|
|||||||||
|
Total non-current assets
|
44,263
|
61,368
|
||||||||||
|
Current assets
|
||||||||||||
|
Trade and other receivables
|
15
|
10,280
|
9,639
|
|||||||||
|
Tax receivable
|
8
|
12,935
|
40,410
|
|||||||||
|
Embedded derivative assets
|
7
|
-
|
266
|
|||||||||
|
Cash and cash equivalents
|
16
|
129,716
|
73,966
|
|||||||||
|
Total current assets
|
152,931
|
124,281
|
||||||||||
|
Total assets
|
197,194
|
185,649
|
||||||||||
|
Equity
|
||||||||||||
|
Share capital
|
17
|
1
|
-
|
|||||||||
|
Share premium
|
17
|
386,230
|
283,250
|
|||||||||
|
Foreign currency translation reserve
|
17
|
163
|
(32
|
)
|
||||||||
|
Share-based payment reserve
|
17, 25
|
18,821
|
10,659
|
|||||||||
|
Accumulated deficit
|
(349,869
|
)
|
(279,106
|
)
|
||||||||
|
Total equity
|
55,346
|
14,771
|
||||||||||
|
Non-current liabilities
|
||||||||||||
|
Interest-bearing loans and borrowings
|
18
|
36,654
|
-
|
|||||||||
|
Deferred liabilities
|
19
|
24,868
|
47,961
|
|||||||||
|
Lease liabilities
|
12
|
25,190
|
38,299
|
|||||||||
|
Provisions
|
20
|
138
|
105
|
|||||||||
|
Total non-current liabilities
|
86,850
|
86,365
|
||||||||||
|
Current liabilities
|
||||||||||||
|
Interest-bearing loans and borrowings
|
21
|
-
|
19,157
|
|||||||||
|
Trade and other payables
|
22
|
25,728
|
29,501
|
|||||||||
|
Deferred liabilities
|
23
|
27,118
|
28,522
|
|||||||||
|
Tax payable
|
24
|
-
|
72
|
|||||||||
|
Lease liabilities
|
12
|
2,043
|
1,951
|
|||||||||
|
Derivative liabilities
|
7
|
-
|
5,127
|
|||||||||
|
Provisions
|
20
|
109
|
183
|
|||||||||
|
Total current liabilities
|
54,998
|
84,513
|
||||||||||
|
Total liabilities
|
141,848
|
170,878
|
||||||||||
|
Total equity and liabilities
|
197,194
|
185,649
|
||||||||||
|
Share
capital
|
Share
premium
|
Foreign
currency
translation
reserve
|
Available-
for-sale-
reserve
|
Share-
Based
Payment
reserve
|
Accumulated
deficit
|
Total
equity
|
||||||||||||||||||||||
|
£
|
’000
|
£
|
’000
|
£
|
’000
|
£
|
’000
|
£
|
’000
|
£
|
’000
|
£
|
’000
|
|||||||||||||||
|
At January 1, 2018
|
—
|
223,986
|
(5
|
)
|
14,962
|
6,812
|
(122,016
|
)
|
123,739
|
|||||||||||||||||||
|
Loss for the year
|
—
|
—
|
—
|
—
|
—
|
(71,630
|
)
|
(71,630
|
)
|
|||||||||||||||||||
|
Reclassification on sale of asset held for sale
|
—
|
—
|
—
|
(18,471
|
)
|
—
|
18,471
|
—
|
||||||||||||||||||||
|
Other comprehensive income
|
—
|
—
|
72
|
3,509
|
125
|
—
|
3,706
|
|||||||||||||||||||||
|
Total comprehensive loss for the year
|
—
|
—
|
72
|
(14,962
|
)
|
125
|
(53,159
|
)
|
(67,924
|
)
|
||||||||||||||||||
|
Issue of share capital
|
—
|
101
|
—
|
—
|
—
|
—
|
101
|
|||||||||||||||||||||
|
Equity-settled share-based payment transactions
|
—
|
—
|
—
|
—
|
666
|
—
|
666
|
|||||||||||||||||||||
|
At December 31, 2018
|
—
|
224,087
|
67
|
—
|
7,603
|
(175,175
|
)
|
56,582
|
||||||||||||||||||||
|
Loss for the year
|
—
|
—
|
—
|
—
|
—
|
(103,931
|
)
|
(103,931
|
)
|
|||||||||||||||||||
|
Other comprehensive loss
|
—
|
—
|
(99
|
)
|
—
|
—
|
—
|
(99
|
)
|
|||||||||||||||||||
|
Total comprehensive loss for the year
|
—
|
—
|
(99
|
)
|
—
|
—
|
(103,931
|
)
|
(104,030
|
)
|
||||||||||||||||||
|
Issue of share capital
|
—
|
59,163
|
—
|
—
|
—
|
—
|
59,163
|
|||||||||||||||||||||
|
Equity-settled share-based payment transactions
|
—
|
—
|
—
|
3,056
|
—
|
3,056
|
||||||||||||||||||||||
|
At December 31, 2019
|
—
|
283,250
|
(32
|
)
|
—
|
10,659
|
(279,106
|
)
|
14,771
|
|||||||||||||||||||
|
Loss for the year
|
—
|
—
|
—
|
—
|
—
|
(74,093
|
)
|
(74,093
|
)
|
|||||||||||||||||||
|
Other comprehensive income
|
—
|
—
|
195
|
—
|
—
|
—
|
195
|
|||||||||||||||||||||
|
Total comprehensive loss for the year
|
—
|
—
|
195
|
—
|
—
|
(74,093
|
)
|
(73,898
|
)
|
|||||||||||||||||||
|
Conversion of interest-bearing loan
|
—
|
—
|
—
|
—
|
—
|
(510
|
)
|
(510
|
)
|
|||||||||||||||||||
|
Derecognition of derivative liability
|
—
|
—
|
—
|
—
|
—
|
3,840
|
3,840
|
|||||||||||||||||||||
|
Issue of share capital
|
1
|
102,980
|
—
|
—
|
—
|
—
|
102,981
|
|||||||||||||||||||||
|
Equity-settled share-based payment transactions
|
—
|
—
|
—
|
—
|
8,162
|
—
|
8,162
|
|||||||||||||||||||||
|
At December 31, 2020
|
1
|
386,230
|
163
|
—
|
18,821
|
(349,869
|
)
|
55,346
|
||||||||||||||||||||
|
Notes
|
2020
£’000
|
2019
£’000
|
2018
£’000
|
|||||||||||||
|
Cash flows from operating activities
|
||||||||||||||||
|
Loss for the year
|
(74,093
|
)
|
(103,931
|
)
|
(71,630
|
)
|
||||||||||
|
Adjustments for:
|
||||||||||||||||
|
Depreciation of property, plant and equipment
|
11
|
6,446
|
6,549
|
6,410
|
||||||||||||
|
Depreciation of right of use assets
|
12
|
2,530
|
2,454
|
-
|
||||||||||||
|
Amortization of intangible assets
|
10
|
-
|
210
|
297
|
||||||||||||
|
Write-off of intangible assets
|
-
|
306
|
170
|
|||||||||||||
|
Loss on disposal of property, plant and equipment
|
11
|
1,064
|
3
|
135
|
||||||||||||
|
Gross gain from sale of equity investment
|
-
|
-
|
(5,204
|
)
|
||||||||||||
|
Profit on derecognition of leases
|
12
|
(3,700
|
)
|
-
|
-
|
|||||||||||
|
Remeasurement of leases
|
12
|
(227
|
)
|
-
|
-
|
|||||||||||
|
Net finance costs/(income)
|
1,167
|
7,867
|
(298
|
)
|
||||||||||||
|
Movement in provisions and other charges
|
20
|
(41
|
)
|
71
|
(50
|
)
|
||||||||||
|
Foreign exchange translation differences
|
(787)
|
(618
|
)
|
1,157
|
||||||||||||
|
Equity settled share-based payment expenses
|
25
|
8,162
|
3,056
|
666
|
||||||||||||
|
Taxation charge
|
8
|
(13,267
|
)
|
(22,258
|
)
|
(16,548
|
)
|
|||||||||
|
Working capital adjustments:
|
||||||||||||||||
|
(Increase) / decrease in trade and other receivables
|
15
|
(532
|
)
|
1,828
|
(1,522
|
)
|
||||||||||
|
(Decrease) / increase in trade and other payables
|
22
|
(3,774
|
)
|
9,946
|
5,300
|
|||||||||||
|
(Decrease)/increase in deferred liabilities
|
19, 23
|
(24,497
|
)
|
(21,866
|
)
|
63,797
|
||||||||||
|
Cash used in operations
|
(101,549
|
)
|
(116,383
|
)
|
(17,320
|
)
|
||||||||||
|
Bank interest received on cash and cash equivalents
|
6
|
676
|
1,525
|
760
|
||||||||||||
|
Net taxation received
|
8
|
40,299
|
13,482
|
(66
|
)
|
|||||||||||
|
Net cash used in operating activities
|
(60,574
|
)
|
(101,376
|
)
|
(16,626
|
)
|
||||||||||
|
Cash flows from investing activities
|
||||||||||||||||
|
Proceeds from sale of property, plant and equipment
|
11
|
675
|
82
|
-
|
||||||||||||
|
Gross proceeds from disposal of equity investment
|
14
|
-
|
-
|
27,451
|
||||||||||||
|
Purchase of property, plant and equipment
|
11
|
(3,074
|
)
|
(4,078
|
)
|
(3,486
|
)
|
|||||||||
|
Purchase of intangible assets
|
10
|
-
|
(198
|
)
|
(51
|
)
|
||||||||||
|
Proceeds from sub-leases
|
12
|
378
|
57
|
-
|
||||||||||||
|
Leasehold incentive
|
13
|
2,488
|
-
|
34,100
|
||||||||||||
|
Net cash flows from / (used) in investing activities
|
467
|
(4,137
|
)
|
58,014
|
||||||||||||
|
Cash flows from financing activities
|
||||||||||||||||
|
Proceeds from exercise of share options
|
25
|
73
|
27
|
101
|
||||||||||||
|
Gross proceeds from issue of share capital
|
17
|
83,218
|
59,874
|
-
|
||||||||||||
|
Costs from issue of share capital
|
17
|
(176
|
)
|
(738
|
)
|
-
|
||||||||||
|
Non-current interest-bearing loan received
|
18
|
37,543
|
-
|
-
|
||||||||||||
|
Interest paid on non-current interest-bearing loan
|
18
|
(291
|
)
|
-
|
-
|
|||||||||||
|
Repayment of lease liabilities
|
12
|
(4,426
|
)
|
(4,036
|
)
|
-
|
||||||||||
|
Net cash flows from financing activities
|
115,941
|
55,127
|
101
|
|||||||||||||
|
Increase / (decrease) in net cash and cash equivalents
|
55,834
|
(50,386
|
)
|
41,489
|
||||||||||||
|
Net foreign exchange difference on cash held
|
(84
|
)
|
(33
|
)
|
13
|
|||||||||||
|
Cash and cash equivalents at beginning of the year
|
73,966
|
124,385
|
82,883
|
|||||||||||||
|
Cash and cash equivalents at end of the year
|
129,716
|
73,966
|
124,385
|
|||||||||||||
| 1. |
Accounting policies
|
|
1.
|
Accounting policies (continued)
|
| • |
determine whether promises contained within the collaboration agreements are distinct from the other promises in the contract;
|
| • |
whether milestones or other variable consideration should be included in the transaction price;
|
| • |
whether performance obligations are satisfied at a point in time or over time, and
|
| • |
for performance obligations satisfied over time the appropriate method of measuring progress for the purposes of revenue recognition.
|
|
1.
|
Accounting policies (continued)
|
| • |
whether achievement of a development milestone is highly susceptible to factors outside the entity’s influence, such as milestones involving the judgment or actions of third parties, including regulatory bodies
or the customer;
|
| • |
whether the uncertainty about the achievement of the milestone is not expected to be resolved for a long period of time;
|
| • |
whether the Company can reasonably predict that a milestone will be achieved based on previous experience; and.
|
| • |
the complexity and inherent uncertainty underlying the achievement of the milestone.
|
|
1.
|
Accounting policies (continued)
|
Under these collaboration agreements, depending on the terms, the Group may also receive commercialization milestones upon the first commercial sale of a product, the amount of which is based on the territory the sale occurs in, and royalties based on worldwide net sales. These amounts have not been included within the transaction price as of December 31, 2020, 2019 and 2018 because they are sales-based royalties which will be recognized when the subsequent sale occurs.
| • |
adjustments arising from a change in the estimate of when the performance obligation will have been completed.
|
| • |
adjustment to revenue that affects deferred revenue;
|
| • |
a change in the estimate of the transaction price due to changes in the assessment of whether variable consideration is constrained because it is not considered probable of being received; and
|
| • |
the recognition of revenue.
|
| • |
past history and experience with similar contracts.
|
| • |
unexpected fluctuations in planned spend.
|
| • |
changes to project timelines.
|
| 1. |
Accounting policies (continued)
|
| • |
the data generated from the Group’s research and development programs;
|
| • |
the future operating performance, prospects and business strategy;
|
|
|
1.
|
Accounting policies (continued)
|
| • |
the material risks related to the Group’s business and industry
|
| • |
the lack of an active public market for the Group’s ordinary and convertible preferred shares;
|
| • |
the market performance of publicly traded companies in the life science and biotechnology sectors;
|
| • |
the prices at which the Group issued ordinary and preferred shares and the superior rights and preferences of the preferred shares relative to the ordinary shares at the time of each grant; and
|
| • |
the likelihood of achieving a liquidity events for the holders of our ordinary shares, series A and B shares and Growth Shares, such as an IPO, given prevailing market conditions.
|
|
|
1.
|
Accounting policies (continued)
|
| 1. |
Accounting policies (continued)
|
| • |
Level 1: quoted prices (unadjusted) in active markets for identical assets or liabilities.
|
| • |
Level 2: inputs other than quoted prices included in Level 1 that are observable for the asset or liability, either directly (i.e. as prices) or indirectly (i.e. derived from prices).
|
|
|
1.
|
Accounting policies (continued)
|
| • |
Level 3: inputs for the asset or liability that are not based on observable market data (unobservable inputs).
|
|
|
2.
|
Revenue & segmental reporting
|
|
2020
£’000
|
2019
£’000
|
2018
£’000
|
||||||||||
|
GlaxoSmithKline
|
6,356
|
5,753
|
6,079
|
|||||||||
|
Eli Lilly
|
3,522
|
819
|
8,561
|
|||||||||
|
Genentech
|
20,236
|
19,097
|
1,461
|
|||||||||
|
MedImmune
|
-
|
-
|
7,553
|
|||||||||
|
30,114
|
25,669
|
23,654
|
||||||||||
|
United Kingdom
|
6,356
|
5,753
|
6,079
|
|||||||||
|
United States
|
23,758
|
19,916
|
17,575
|
|||||||||
|
30,114
|
25,669
|
23,654
|
|
At 1 January
2020
£’000
|
Additions
£’000
|
Deductions
£’000
|
At December
31, 2020
£’000
|
|||||||||||||
|
Trade receivables:
|
||||||||||||||||
|
Trade receivables
|
1,186
|
4,023
|
(5,209
|
)
|
-
|
|||||||||||
|
Total receivables
|
1,186
|
4,023
|
(5,209
|
)
|
-
|
|||||||||||
|
Contract assets:
|
||||||||||||||||
|
Contract assets
|
424
|
1,658
|
-
|
2,082
|
||||||||||||
|
Total contract assets
|
424
|
1,658
|
-
|
2,082
|
||||||||||||
|
Contract liabilities:
|
||||||||||||||||
|
Deferred revenue
|
76,418
|
-
|
(24,432
|
)
|
51,986
|
|||||||||||
|
Total contract liabilities
|
76,418
|
-
|
(24,432
|
)
|
51,986
|
|||||||||||
|
At 1
January
2019
£’000
|
Additions
£’000
|
Deductions
£’000
|
At
December 31,
2019
£’000
|
|||||||||||||
|
Trade receivables:
|
||||||||||||||||
|
Trade receivables
|
3,600
|
3,431
|
(5,845
|
)
|
1,186
|
|||||||||||
|
Total receivables
|
3,600
|
3,431
|
(5,845
|
)
|
1,186
|
|||||||||||
|
Contract assets:
|
||||||||||||||||
|
Contract assets
|
-
|
424
|
-
|
424
|
||||||||||||
|
Total contract assets
|
-
|
424
|
-
|
424
|
||||||||||||
|
Contract liabilities:
|
||||||||||||||||
|
Deferred revenue
|
98,232
|
-
|
(21,814
|
)
|
76,418
|
|||||||||||
|
Total contract liabilities
|
98,232
|
-
|
(21,814
|
)
|
76,418
|
|||||||||||
|
2020
£’000
|
2019
£’000
|
|||||||
|
Current deferred revenue (Note 23)
|
27,118
|
28,457
|
||||||
|
Non-current deferred revenue (Note 19)
|
24,868
|
47,961
|
||||||
|
51,986
|
76,418
|
|||||||
|
3.
|
Operating loss is stated after charging:
|
|
2020
£’000
|
2019
£’000
|
2018
£’000
|
||||||||||
|
Research and development costs
|
74,809
|
99,991
|
83,575
|
|||||||||
|
Loss on disposal of property, plant and equipment
|
1,064
|
3
|
135
|
|||||||||
|
Profit on derecognition of leases (Note 12)
|
(3,700
|
)
|
-
|
-
|
||||||||
|
Remeasurement of leases (Note 12)
|
(227
|
)
|
-
|
-
|
||||||||
|
Loss on write-offs of intangible fixed assets
|
-
|
306
|
170
|
|||||||||
|
Depreciation of property, plant and equipment (Note 11)
|
6,446
|
9,003
|
6,410
|
|||||||||
|
Amortization of intangible assets (Note 10)
|
-
|
210
|
297
|
|||||||||
|
Operating lease expense (Note 12)
|
296
|
486
|
4,205
|
|||||||||
|
Operating lease income (Note 5)
|
460
|
185
|
(622
|
)
|
||||||||
|
Realized foreign exchange (gains)/loss
|
477
|
189
|
(1,341
|
)
|
||||||||
|
4.
|
Staff numbers and costs
|
|
2020
No. of employees
|
2019
No. of employees
|
2018
No. of employees
|
||||||||||
|
Research
|
177
|
284
|
299
|
|||||||||
|
Development
|
96
|
108
|
95
|
|||||||||
|
Corporate
|
56
|
67
|
67
|
|||||||||
|
Total
|
329
|
459
|
461
|
|||||||||
|
The aggregate staff costs of these persons were as follows:
|
2020
£’000
|
2019
£’000
|
2018
£’000
|
|||||||||
|
Wages and salaries
|
29,038
|
31,920
|
29,501
|
|||||||||
|
Social security costs
|
2,131
|
2,767
|
2,731
|
|||||||||
|
Share-based payments (Note 25)
|
8,162
|
3,056
|
666
|
|||||||||
|
Contributions to defined contribution plans (Note 27)
|
1,035
|
1,213
|
981
|
|||||||||
|
40,366
|
38,956
|
33,879
|
||||||||||
| 5. |
Net other operating income
|
|
2020
£’000
|
2019
£’000
|
2018
£’000
|
||||||||||
|
Profit on derecognition of leases
|
3,700
|
-
|
||||||||||
|
Loss on disposal of property, plant and equipment
|
(1,064
|
)
|
-
|
|||||||||
|
Settlement agreement
|
810
|
-
|
||||||||||
|
Sub-lease income
|
460
|
185
|
622
|
|||||||||
|
Remeasurement of leases
|
227
|
|||||||||||
|
Other
|
109
|
-
|
||||||||||
|
4,242
|
185
|
622
|
||||||||||
|
6.
|
Finance income
|
|
2020
£’000
|
2019
£’000
|
2018
£’000
|
||||||||||
|
Bank interest on cash and cash equivalents
|
668
|
1,386
|
550
|
|||||||||
|
Interest on short-term deposits
|
-
|
-
|
272
|
|||||||||
|
Gain on entering into sub-leases on leasehold properties
|
215
|
115
|
-
|
|||||||||
|
Interest on investment in sub-lease
|
38
|
9
|
-
|
|||||||||
|
Gain from change in fair value of derivative liability
|
1,287
|
-
|
-
|
|||||||||
|
Gain from change in fair value of derivative asset
|
-
|
-
|
318
|
|||||||||
|
2,208
|
1,510
|
1,140
|
||||||||||
|
7.
|
Finance costs
|
|
2020
£’000
|
2019
£’000
|
2018
£’000
|
||||||||||
|
Interest on lease liabilities
|
2,401
|
2,947
|
-
|
|||||||||
|
Interest expenses on financial liabilities measured at amortized cost
|
708
|
849
|
842
|
|||||||||
|
Loss from change in fair value of embedded derivative asset
|
266
|
454
|
-
|
|||||||||
|
Loss from change in fair value of derivative liability
|
-
|
5,127
|
-
|
|||||||||
|
Other finance costs
|
-
|
2
|
-
|
|||||||||
|
3,375
|
9,379
|
842
|
||||||||||
|
7.
|
Finance costs (continued)
|
|
8.
|
Income tax
|
|
2020
£’000
|
2019
£’000
|
2018
£’000
|
||||||||||
|
Profit or loss
|
||||||||||||
|
Current tax:
|
||||||||||||
|
R&D tax credit for the year
|
(12,432
|
)
|
(21,767
|
)
|
(18,486
|
)
|
||||||
|
Tax related to share-based compensation plans
|
-
|
-
|
125
|
|||||||||
|
Foreign corporation tax on profits for the year
|
84
|
152
|
139
|
|||||||||
|
Adjustments in respect of prior years
|
(100
|
)
|
43
|
-
|
||||||||
|
Total current tax
|
(12,448
|
)
|
(21,572
|
)
|
(18,222
|
)
|
||||||
|
Deferred tax:
|
||||||||||||
|
Current year
|
(790
|
)
|
-
|
-
|
||||||||
|
Effect of changes in tax rates
|
(1
|
)
|
-
|
-
|
||||||||
|
Movement in unrecognized deferred tax asset
|
351
|
-
|
-
|
|||||||||
|
Originating and reversal of timing differences, including adjustments in respect of prior years
|
(379
|
)
|
(686
|
)
|
1,674
|
|||||||
|
Total deferred tax
|
(819
|
)
|
(686
|
)
|
1,674
|
|||||||
|
Total income tax credit
|
(13,267
|
)
|
(22,258
|
)
|
(16,548
|
)
|
||||||
|
Tax related to items recognized in other comprehensive income during the year:
|
||||||||||||
|
Current tax related to share-based compensation plans
|
-
|
-
|
(125
|
)
|
||||||||
|
Deferred tax on fair value movements of available-for-sale financial assets
|
-
|
-
|
(3,509
|
)
|
||||||||
|
Tax charged to other comprehensive income
|
-
|
-
|
(3,634
|
)
|
||||||||
|
8.
|
Income tax (continued)
|
|
Reconciliation of tax expense and accounting profit for 2020, 2019 and 2018:
|
2020
|
2019
|
2018
|
|||||||||
|
£
|
’000
|
£
|
’000
|
£
|
’000
|
|||||||
|
Loss before tax
|
(87,360
|
)
|
(126,189
|
)
|
(88,178
|
)
|
||||||
|
Tax credit using the UK Corporation tax rate of 19% (2019: 19% and 2018: 19%)
|
(16,598
|
)
|
(23,976
|
)
|
(16,754
|
)
|
||||||
|
Effect of:
|
||||||||||||
|
Non-deductible expenses
|
9,120
|
13,148
|
629
|
|||||||||
|
Income not taxable for tax purposes
|
-
|
-
|
(954
|
)
|
||||||||
|
Chargeable gain on sale of assets held for sale
|
-
|
-
|
4,359
|
|||||||||
|
Other permanent differences
|
-
|
(1
|
)
|
(38
|
)
|
|||||||
|
Additional deduction for R&D expenditure
|
(16,286
|
)
|
(29,365
|
)
|
(13,691
|
)
|
||||||
|
Surrender of tax losses for R&D tax credit refund
|
16,286
|
28,523
|
24,223
|
|||||||||
|
R&D expenditure credits
|
(13,424
|
)
|
(22,602
|
)
|
(19,215
|
)
|
||||||
|
Credit to other comprehensive income for share-based compensation plans
|
-
|
-
|
125
|
|||||||||
|
Movement in deferred tax not recognized
|
8,084
|
12,413
|
4,746
|
|||||||||
|
Adjustments to tax charge in respect of previous periods - deferred tax
|
(379
|
)
|
(500
|
)
|
-
|
|||||||
|
Adjustments to tax charge in respect of previous periods
|
(100
|
)
|
43
|
-
|
||||||||
|
State taxes
|
7
|
-
|
-
|
|||||||||
|
Effects of overseas tax rates
|
24
|
-
|
-
|
|||||||||
|
Effects of tax rates in foreign jurisdictions
|
(1
|
)
|
59
|
22
|
||||||||
|
Total tax credit included in loss for the year
|
(13,267
|
)
|
(22,258
|
)
|
(16,548
|
)
|
||||||
|
2020
£’000
|
2019
£’000
|
2018
£’000
|
||||||||||
|
Current tax:
|
||||||||||||
|
United States:
|
||||||||||||
|
Federal
|
(16
|
)
|
100
|
137
|
||||||||
|
State
|
(-
|
)
|
15
|
2
|
||||||||
|
United Kingdom
|
(12,432
|
)
|
(21,687
|
)
|
(18,361
|
)
|
||||||
|
Total current tax
|
(12,448
|
)
|
(21,572
|
)
|
(18,222
|
)
|
||||||
|
Deferred tax:
|
||||||||||||
|
United States:
|
||||||||||||
|
Federal
|
(819
|
)
|
(644
|
)
|
(516
|
)
|
||||||
|
State
|
-
|
(42
|
)
|
(1
|
)
|
|||||||
|
United Kingdom
|
-
|
-
|
2,191
|
|||||||||
|
Total deferred tax
|
(819
|
)
|
(686
|
)
|
1,674
|
|||||||
|
Total income tax credit
|
(13,267
|
))
|
(22,258
|
)
|
(16,548
|
)
|
||||||
|
8.
|
Income tax (continued)
|
|
2020
£’000
|
2019
£’000
|
2018
£’000
|
||||||||||
|
United States
|
—
|
—
|
—
|
|||||||||
|
United Kingdom – current tax
|
—
|
—
|
(125
|
)
|
||||||||
|
United Kingdom – deferred tax
|
—
|
—
|
(3,509
|
)
|
||||||||
|
Tax charged to other comprehensive income
|
—
|
—
|
(3,634
|
)
|
||||||||
|
2020
|
2019
|
2018
|
||||||||||
|
Loss for the year (£000's)
|
(74,093
|
)
|
(103,931
|
)
|
(71,630
|
)
|
||||||
|
Basic and diluted weighted average number of shares
|
26,523,411
|
22,297,935
|
21,558,890
|
|||||||||
|
Basic and diluted loss per share (£) (1)
|
(2.79
|
)
|
(4.66
|
)
|
(3.32
|
)
|
||||||
|
(1)
|
The basic and diluted loss per share are adjusted for the (i) the exchange of shares of Immunocore Limited for shares of
Immunocore Holdings Limited on a 1 for 100 basis, and (ii) the reorganization of the share capital of Immunocore Holdings plc, resulting in a consolidation with the effect of a 20 to 1 reverse stock split on the Company’s ordinary
shares and non-voting ordinary shares, all of which took place in connection with the Company’s initial public offering which closed on February 9, 2021. No other adjustments have been made to the consolidated financial statements
of the Group in regard to the corporate reorganization. Refer to Note 30 for further information.
|
|
10.
|
Intangible assets
|
|
Patents £'000
|
Computer
software £’000
|
Assets under
construction
£’000
|
Total £’000
|
|||||||||||||
|
Cost:
|
||||||||||||||||
|
At January 1, 2019
|
516
|
867
|
13
|
1,396
|
||||||||||||
|
Additions
|
-
|
76
|
122
|
198
|
||||||||||||
|
Transferred
|
-
|
24
|
(24
|
)
|
-
|
|||||||||||
|
Write-offs
|
-
|
(967
|
)
|
(111
|
)
|
(1,078
|
)
|
|||||||||
|
At December 31, 2019
|
516
|
-
|
-
|
516
|
||||||||||||
|
Additions
|
-
|
-
|
-
|
-
|
||||||||||||
|
Transferred
|
-
|
-
|
-
|
-
|
||||||||||||
|
Write-offs
|
-
|
-
|
-
|
-
|
||||||||||||
|
At December 31, 2020
|
516
|
-
|
-
|
516
|
||||||||||||
|
Amortization and impairment:
|
||||||||||||||||
|
At January 1, 2019
|
516
|
562
|
-
|
1,078
|
||||||||||||
|
Write-offs
|
-
|
(772
|
)
|
-
|
(772
|
)
|
||||||||||
|
Amortization for the year
|
-
|
210
|
-
|
210
|
||||||||||||
|
At December 31, 2019
|
516
|
-
|
-
|
516
|
||||||||||||
|
Write-offs
|
-
|
-
|
-
|
-
|
||||||||||||
|
Amortization for the year
|
-
|
-
|
-
|
-
|
||||||||||||
|
At December 31, 2020
|
516
|
-
|
-
|
516
|
||||||||||||
|
Carrying value:
|
||||||||||||||||
|
At December 31, 2020
|
-
|
-
|
-
|
-
|
||||||||||||
|
At December 31, 2019
|
-
|
-
|
-
|
-
|
||||||||||||
|
At January 1, 2019
|
-
|
305
|
13
|
318
|
||||||||||||
| 11. |
Property, plant and equipment
|
|
Leasehold
properties and improvements
£’000
|
Plant and
equipment
£’000
|
Assets under
construction
£’000 |
Total
£’000
|
|||||||||||||
|
Cost:
|
||||||||||||||||
|
At January 1, 2019
|
11,137
|
25,639
|
989
|
37,765
|
||||||||||||
|
Additions
|
215
|
1,150
|
2,713
|
4,078
|
||||||||||||
|
Transfers
|
1,090
|
41
|
(1,131
|
)
|
-
|
|||||||||||
|
Effect of foreign currency translation
|
(17
|
)
|
(4
|
)
|
-
|
(21
|
)
|
|||||||||
|
Disposals
|
(185
|
)
|
(500
|
)
|
-
|
(685
|
)
|
|||||||||
|
At December 31, 2019
|
12,240
|
26,326
|
2,571
|
41,137
|
||||||||||||
|
Additions
|
564
|
775
|
1,735
|
3,074
|
||||||||||||
|
Transfers
|
4,123
|
2
|
(4,125
|
)
|
-
|
|||||||||||
|
Effect of foreign currency translation
|
(27
|
)
|
(2
|
)
|
-
|
(29
|
)
|
|||||||||
|
Disposals
|
(1,090
|
)
|
(1,118
|
)
|
(61
|
)
|
(2,269
|
)
|
||||||||
|
At December 31, 2020
|
15,810
|
25,983
|
120
|
41,913
|
||||||||||||
|
Depreciation and impairment:
|
||||||||||||||||
|
At January 1, 2019
|
3,752
|
13,139
|
-
|
16,891
|
||||||||||||
|
Depreciation charge for the year
|
2,047
|
4,502
|
-
|
6,549
|
||||||||||||
|
Effect of foreign currency translation
|
(2
|
)
|
(3
|
)
|
-
|
(5
|
)
|
|||||||||
|
Disposals
|
(155
|
)
|
(445
|
)
|
-
|
(600
|
)
|
|||||||||
|
At December 31, 2019
|
5,642
|
17,193
|
-
|
22,835
|
||||||||||||
|
Depreciation charge for the year
|
2,356
|
4,090
|
-
|
6,446
|
||||||||||||
|
Effect of foreign currency translation
|
(7
|
)
|
(67
|
)
|
-
|
(74
|
)
|
|||||||||
|
Disposals
|
(156
|
)
|
(892
|
)
|
-
|
(1,048
|
)
|
|||||||||
|
At December 31, 2020
|
7,835
|
20,324
|
-
|
28,159
|
||||||||||||
|
Carrying value:
|
||||||||||||||||
|
At December 31, 2020
|
7,975
|
5,659
|
120
|
13,754
|
||||||||||||
|
At December 31, 2019
|
6,698
|
9,133
|
2,571
|
18,302
|
||||||||||||
|
At January 1, 2019
|
7,385
|
12,500
|
989
|
20,874
|
||||||||||||
| 12. |
Leases
|
| • |
Options to terminate the lease early at the right of the tenant
|
| • |
Variable lease payments with a guaranteed minimum increase and capped maximum increase
|
|
2020
|
2019
|
|||||||
|
£
|
’000
|
£
|
’000
|
|||||
|
Balance at 1 January
|
36,578
|
-
|
||||||
|
Effect of adopting new accounting standards
|
(31
|
)
|
44,984
|
|||||
|
Additions
|
453
|
897
|
||||||
|
Remeasurements
|
(2,269
|
)
|
(6,849
|
)
|
||||
|
Derecognition
|
(9,108
|
)
|
-
|
|||||
|
Depreciation charge for the year
|
(2,530
|
)
|
(2,454
|
)
|
||||
|
23,093
|
36,578
|
|||||||
|
2020
£’000
|
2019
£’000
|
|||||||
|
Less than one year
|
3,560
|
4,469
|
||||||
|
One to five years
|
9,607
|
16,834
|
||||||
|
More than five years
|
32,600
|
45,288
|
||||||
|
Total undiscounted lease liabilities
|
45,767
|
66,591
|
||||||
| 12. |
Leases (continued)
|
|
2020
£’000
|
2019
£’000
|
|||||||
|
Current
|
2,043
|
1,951
|
||||||
|
Non-current
|
25,190
|
38,299
|
||||||
|
Total lease liabilities
|
27,233
|
40,250
|
||||||
|
Amounts recognized in the Consolidated Statements of Loss
|
2020
£’000
|
2019
£’000
|
||||||
|
Interest on lease liabilities
|
2,401
|
2,947
|
||||||
|
Expenses relating to short-term leases
|
296
|
486
|
||||||
|
Expenses relating to leases of low-value assets
|
19
|
33
|
||||||
|
Interest on investment in sub-lease
|
(38
|
)
|
(9
|
)
|
||||
|
2020
£’000
|
2019
£000’s
|
2018
£’000’s
|
||||||||||
|
Within one year
|
-
|
73
|
4,329
|
|||||||||
|
After one year but not more than five years
|
-
|
-
|
16,566
|
|||||||||
|
More than five years
|
-
|
-
|
60,691
|
|||||||||
|
-
|
73
|
81,586
|
||||||||||
|
Amounts recognized in the Consolidated Statement of Cash Flows
|
2020
£’000s
|
2019
£’000
|
||||||
|
Total cash outflow for leases
|
4,426
|
4,036
|
||||||
|
Lease income
|
2020
£’000
|
2019
£’000
|
||||||
|
Operating lease income
|
460
|
185
|
||||||
|
Finance lease income on the net investment in the lease
|
38
|
9
|
||||||
| 12. |
Leases (continued)
|
|
Maturity analysis – undiscounted finance lease income
|
2020
£’000
|
2019
£’000
|
||||||
|
Less than one year
|
720
|
318
|
||||||
|
One to two years
|
96
|
300
|
||||||
|
Two to three years
|
-
|
12
|
||||||
|
Three to four years
|
-
|
-
|
||||||
|
Four to five years
|
-
|
-
|
||||||
|
More than five years
|
-
|
-
|
||||||
|
Total undiscounted finance lease income
|
816
|
630
|
||||||
|
Unearned finance income
|
(40
|
)
|
(39
|
)
|
||||
|
Net investment in the lease
|
776
|
591
|
||||||
|
Maturity analysis – undiscounted operating lease income
|
2020
£’000
|
2019
£’000
|
2018
£’000
|
|||||||||
|
Less than one year
|
-
|
96
|
176
|
|||||||||
|
One to two years
|
-
|
50
|
11
|
|||||||||
|
Two to three years
|
-
|
12
|
11
|
|||||||||
|
Three to four years
|
-
|
-
|
11
|
|||||||||
|
Four to five years
|
-
|
-
|
11
|
|||||||||
|
More than five years
|
-
|
-
|
-
|
|||||||||
|
Total undiscounted operating lease income
|
-
|
158
|
220
|
|||||||||
| 13. |
Other non-current financial assets
|
|
2020
£’000
|
2019
£’000
|
|||||||
|
Long-term security deposits
|
786
|
2,532
|
||||||
|
Prepayments
|
3,427
|
1,858
|
||||||
|
Other
|
197
|
-
|
||||||
|
4,410
|
4,390
|
|||||||
| 14. |
Available for sale assets
|
| 15. |
Trade and other receivables
|
|
2020
£’000
|
2019
£’000
|
|||||||
|
Trade receivables
|
2,051
|
1,471
|
||||||
|
Other receivables
|
1,722
|
3,667
|
||||||
|
Interest receivable
|
-
|
28
|
||||||
|
Prepayments and accrued income
|
6,507
|
4,473
|
||||||
|
10,280
|
9,639
|
|||||||
| 16. |
Cash and cash equivalents
|
|
2020
£’000
|
2019
£000’s
|
|||||||
|
Cash at bank and in hand
|
129,716
|
73,966
|
||||||
|
129,716
|
73,966
|
|||||||
| 17. |
Capital and reserves
|
|
|
Growth
shares
|
Series A
shares
|
Series B
shares
|
Series C
shares
|
Ordinary
shares
|
|||||||||||||||
|
Issued share capital
(0.01p per share)
|
||||||||||||||||||||
|
At January 1, 2018
|
155,246
|
1,699,576
|
-
|
-
|
2,459,363
|
|||||||||||||||
|
New shares issued for cash
|
-
|
-
|
10,950
|
|||||||||||||||||
|
Repurchased and cancelled
|
(36,800
|
)
|
-
|
-
|
-
|
-
|
||||||||||||||
|
At January 1, 2019
|
118,446
|
1,699,576
|
-
|
-
|
2,470,313
|
|||||||||||||||
|
New shares issued for cash
|
-
|
-
|
621,556
|
-
|
45,581
|
|||||||||||||||
|
Repurchased and cancelled
|
(60,240
|
)
|
-
|
-
|
-
|
-
|
||||||||||||||
|
At December 31, 2019
|
58,206
|
1,699,576
|
621,556
|
-
|
2,515,894
|
|||||||||||||||
|
New shares issued for cash
|
34,260
|
-
|
323,450
|
823,719
|
163,870
|
|||||||||||||||
|
New shares issued for non-cash consideration
|
- | - |
203,697
|
- | - | |||||||||||||||
|
Repurchased and cancelled
|
(29,575
|
)
|
-
|
-
|
-
|
-
|
||||||||||||||
|
At December 31, 2020
|
62,891
|
1,699,576
|
1,148,703
|
823,719
|
2,679,764
|
|||||||||||||||
|
2020
£
|
2019
£
|
2018
£
|
||||||||||
|
Allotted, called up and fully paid
|
||||||||||||
|
Ordinary shares
|
268
|
252
|
247
|
|||||||||
|
Series A shares
|
170
|
170
|
170
|
|||||||||
|
Series B shares
|
115
|
62
|
-
|
|||||||||
|
Series C shares
|
82
|
-
|
-
|
|||||||||
|
Growth shares
|
6
|
6
|
12
|
|||||||||
|
641
|
490
|
429
|
||||||||||
| 17. |
Capital and reserves (continued)
|
|
Share premium
|
||||
|
£
|
’000
|
|||
|
At January 1, 2018
|
223,986
|
|||
|
New shares issued for cash
|
101
|
|||
|
At December 31, 2018
|
224,087
|
|||
|
New shares issued for cash
|
59,163
|
|||
|
At December 31, 2019
|
283,250
|
|||
|
New shares issued for cash
|
83,115
|
|||
|
New shares issued for non-cash consideration
|
19,865
|
|||
|
At December 31, 2020
|
386,230
|
|||
| • |
managing the budgeting process;
|
| • |
managing funding and liquidity risk; and
|
| • |
maintaining strong investor relations
|
| 18. |
Non-current interest-bearing loans and borrowings
|
|
2020
£’000
|
2019
£’000
|
|||||||
|
Long-term borrowings
|
36,654
|
-
|
||||||
|
36,654
|
-
|
|||||||
| 19. |
Non-Current Deferred liabilities
|
|
2020
£’000
|
2019
£’000
|
|||||||
|
Deferred revenue
|
24,868
|
47,961
|
||||||
|
24,868
|
47,961
|
|||||||
| 20. |
Provisions
|
|
Total
£’000
|
||||
|
At January 1, 2019
|
217
|
|||
|
Arising during the year
|
150
|
|||
|
Utilized
|
(79
|
)
|
||
|
At December 31, 2019
|
288
|
|||
|
Arising during the year
|
299
|
|||
|
Utilized
|
(340
|
)
|
||
|
At December 31, 2020
|
247
|
|||
|
Current
|
109
|
|||
|
Non-current
|
138
|
|||
| 21. |
Current interest-bearing loans and borrowings
|
|
2020
£’000
|
2019
£000’s
|
|||||||
|
Short-term convertible loan (Note 26)
|
-
|
19,157
|
||||||
|
-
|
19,157
|
|||||||
| 21. |
Current interest-bearing loans and borrowings (continued)
|
| 22. |
Trade and other payables
|
|
2020
£’000
|
2019
£’000
|
|||||||
|
Trade payables
|
5,783
|
15,729
|
||||||
|
Other taxation and social security
|
620
|
522
|
||||||
|
Pension Liability
|
2
|
1
|
||||||
|
Accruals
|
19,323
|
13,249
|
||||||
|
25,728
|
29,501
|
|||||||
| 23. |
Current deferred liabilities
|
|
2020
£’000
|
2019
£’000
|
|||||||
|
Deferred revenue
|
27,118
|
28,457
|
||||||
|
Deferred rent
|
-
|
65
|
||||||
|
27,118
|
28,522
|
|||||||
| 24. |
Tax payable
|
|
2020
£’000
|
2019
£’000
|
|||||||
|
Tax payable
|
-
|
72
|
||||||
|
-
|
72
|
|||||||
| 25. |
Share-based payments
|
| 25. |
Share-based payments (continued)
|
|
Number of shares issuable
|
Number of share
options (#)
|
Weighted average
exercise price (£)
|
||||||
|
Outstanding at January 1, 2018
|
227,608
|
54.01
|
||||||
|
Awards granted
|
-
|
-
|
||||||
|
Awards exercised
|
(10,950
|
)
|
9.26
|
|||||
|
Awards forfeited
|
(67,935
|
)
|
53.57
|
|||||
|
Outstanding at December 31, 2018
|
148,723
|
57.50
|
||||||
|
Awards granted
|
582,252
|
150.00
|
||||||
|
Awards exercised
|
(8,574
|
)
|
2.71
|
|||||
|
Awards forfeited
|
(6,578
|
)
|
103.17
|
|||||
|
Outstanding at December 31, 2019
|
715,823
|
132.89
|
||||||
|
Awards granted
|
224,536
|
64.00
|
||||||
|
Awards exercised
|
(2,776
|
)
|
21.91
|
|||||
|
Awards forfeited
|
(27,311
|
)
|
81.67
|
|||||
|
Outstanding at December 31, 2020
|
910,272
|
62.90
|
||||||
|
Exercisable at December 31, 2020
|
194,106
|
91.92
|
||||||
| 25. |
Share-based payments (continued)
|
|
Number of shares issuable
|
Number of
growth shares
|
Weighted average
hurdle rate £
|
||||||
|
Outstanding at January 1, 2018
|
155,246
|
170.00
|
||||||
|
Awards granted
|
-
|
-
|
||||||
|
Awards exercised
|
-
|
-
|
||||||
|
Awards forfeited
|
(36,800
|
)
|
170.00
|
|||||
|
Outstanding at December 31, 2018
|
118,446
|
170.00
|
||||||
|
Awards granted
|
-
|
-
|
||||||
|
Awards exercised
|
-
|
-
|
||||||
|
Awards forfeited
|
(60,240
|
)
|
170.00
|
|||||
|
Outstanding at December 31, 2019
|
58,206
|
170.00
|
||||||
|
Awards granted
|
34,260
|
110.41
|
||||||
|
Awards exercised
|
-
|
-
|
||||||
|
Awards forfeited
|
(29,575
|
)
|
170.00
|
|||||
|
Outstanding at December 31, 2020
|
62,891
|
137.54
|
||||||
|
Exercisable at December 31, 2020
|
34,857
|
157.83
|
||||||
|
Growth Shares
|
Share options
|
||||||||||||||||||||||
|
Hurdle rate £
|
Number of
options
|
Weighted average
remaining
contractual life
|
Exercise price £
|
Number of
options
|
Weighted average
remaining contractual life |
||||||||||||||||||
|
170.00
|
43,631
|
7.3
|
43.37
|
91,994
|
4.7
|
||||||||||||||||||
|
64.00
|
19,260
|
9.4
|
120.87
|
3,309
|
5.0
|
||||||||||||||||||
|
-
|
-
|
-
|
150.00
|
11,481
|
6.3
|
||||||||||||||||||
|
-
|
-
|
-
|
64.00
|
803,488
|
9.5
|
||||||||||||||||||
|
Growth shares
|
Share options
|
|||||||||||||||||||||||
|
Apr-20
|
Jun-20
|
Nov- 20
|
Oct- 20
|
Jun-20
|
Apr- 20
|
|||||||||||||||||||
|
Share price at grant date
|
£
|
64.00
|
£
|
64.00
|
£
|
64.00
|
£
|
64.00
|
£
|
64.00
|
£
|
64.00
|
||||||||||||
|
Exercise price
|
-
|
-
|
£
|
64.00
|
£
|
64.00
|
£
|
64.00
|
£
|
64.00
|
||||||||||||||
|
Hurdle rate
|
£
|
64.00 - £170.00
|
£
|
64.00
|
-
|
-
|
-
|
-
|
||||||||||||||||
|
Expected volatility
|
91
|
%
|
102
|
%
|
87
|
%
|
87
|
%
|
85
|
%
|
79
|
%
|
||||||||||||
|
Expected life (years)
|
1 yr
|
1 yr
|
3 yrs
|
3 yrs
|
3 yrs
|
3 yrs
|
||||||||||||||||||
|
Risk free rate
|
0.03
|
%
|
(0.02
|
)%
|
(0.01
|
)%
|
(0.07
|
)%
|
(0.03)%–0.02
|
%
|
0.03
|
%
|
||||||||||||
|
Fair value
|
£
|
2.12 - £7.05
|
£
|
7.05
|
£
|
35.00
|
£
|
35.16
|
£
|
34.32 - £34.30
|
£
|
32.394
|
||||||||||||
| 25. |
Share-based payments (continued)
|
|
Growth shares
|
Share options
|
Share options
|
Share options
|
|||||||||||||
|
|
Apr-17
|
May-19
|
Apr-17
|
2016
|
||||||||||||
|
Share price at grant date
|
£
|
150.00
|
£
|
64.00
|
£
|
150.00
|
£
|
140.00
|
||||||||
|
Exercise price
|
—
|
£
|
150.00
|
£
|
150.00
|
£
|
43.37 - £150.00
|
|||||||||
|
Hurdle rate
|
£
|
170.00
|
—
|
—
|
—
|
|||||||||||
|
Expected volatility
|
65
|
%
|
67
|
%
|
65
|
%
|
60
|
%
|
||||||||
|
Expected life (years)
|
2.7 yrs
|
1.9 yrs - 3 yrs
|
5 yrs
|
5 yrs
|
||||||||||||
|
Risk free rate
|
0.15
|
%
|
0.69% - 0.71
|
%
|
0.42
|
%
|
0.62% - 1.41
|
%
|
||||||||
|
Fair value
|
£
|
58.55
|
£
|
11.95
|
£
|
80.63
|
£
|
77.16 - £107.94
|
||||||||
| 26. |
Financial instruments
|
| 26. |
Financial instruments (continued)
|
|
At December 31, 2020
|
Carrying
amount
£’000
|
Contractual
cash flows
£’000
|
One year
or less
£’000
|
|||||||||
|
Financial assets
|
||||||||||||
|
Trade receivables
|
1,797
|
1,797
|
1,797
|
|||||||||
|
Clinical trial deposits in current assets
|
1,221
|
1,221
|
1,221
|
|||||||||
|
Non-current financial assets
|
3.573
|
3,573
|
-
|
|||||||||
|
Cash and cash equivalents
|
129,716
|
129,716
|
129,716
|
|||||||||
|
Total financial assets
|
136,307
|
136,307
|
132,734
|
|||||||||
|
Financial liabilities
|
||||||||||||
|
Trade payables
|
25,084
|
25,084
|
25,084
|
|||||||||
|
Interest-bearing loans and borrowings (Note 18)
|
36,654
|
51,421
|
3,354
|
|||||||||
|
Total financial liabilities
|
61,738
|
76,505
|
28,438
|
|||||||||
|
At December 31, 2019
|
Carrying
amount
£’000
|
Contractual
cash flows
£’000
|
One year
or less
£’000
|
|||||||||
|
Financial assets
|
||||||||||||
|
Trade receivables
|
1,471
|
1,471
|
1,471
|
|||||||||
|
Interest receivable
|
28
|
28
|
28
|
|||||||||
|
Prepayments and accrued income
|
2,282
|
2,282
|
424
|
|||||||||
|
Long-term security deposit
|
2,532
|
2,532
|
-
|
|||||||||
|
Cash and cash equivalents
|
73,966
|
73,966
|
73,966
|
|||||||||
|
Total financial assets
|
80,279
|
80,279
|
75,889
|
|||||||||
|
Financial liabilities
|
||||||||||||
|
Trade payables
|
15,579
|
15,579
|
15,579
|
|||||||||
|
Interest-bearing loans and borrowings (Note 21)
|
19,157
|
19,426
|
19,157
|
|||||||||
|
Derivative liability
|
5,127
|
-
|
5,127
|
|||||||||
|
Total financial liabilities
|
39,863
|
35,005
|
39,863
|
|||||||||
| 26. |
Financial instruments (continued)
|
|
2020
Carrying
amount
£’000
|
2019
Carrying
amount
£’000
|
|||||||
|
Cash and cash equivalents
|
129,716
|
73,966
|
||||||
|
129,716
|
73,966
|
|||||||
|
2020
Carrying amount
£’000
|
2019
Carrying amount
£’000
|
|||||||
|
Interest-bearing loans and borrowings
|
36,654
|
19,157
|
||||||
|
36,654
|
19,157-
|
|||||||
| 26. |
Financial instruments (continued)
|
|
2020
Carrying
amount
£’000
|
2019
Carrying
amount
£’000
|
|||||||
|
Financial assets at amortized cost:
|
||||||||
|
Interest receivable
|
-
|
15
|
||||||
|
Clinical trial deposits and other debtors
|
4,036
|
1,858
|
||||||
|
Cash and cash equivalents
|
92,844
|
12,518
|
||||||
|
96,880
|
14,391
|
|||||||
|
Financial liabilities at amortized cost:
|
||||||||
|
Trade payables
|
13,779
|
4,374
|
||||||
|
Interest-bearing loans and borrowings (Notes 18 and 21)
|
36,654
|
19,157
|
||||||
|
50,433
|
23,531
|
|||||||
| 26. |
Financial instruments (continued)
|
|
2020
|
2019
|
|||||||||||||||
|
Carrying amount
£’000
|
Fair value £’000
|
Carrying amount
£’000
|
Fair value £’000
|
|||||||||||||
|
Financial assets at amortized cost:
|
||||||||||||||||
|
Trade receivables
|
1,797
|
1,797
|
1,471
|
1,471
|
||||||||||||
|
Interest receivable
|
-
|
28
|
28
|
|||||||||||||
|
Current clinical trial deposits and accrued income
|
1,221
|
1,221
|
424
|
424
|
||||||||||||
|
Non-current financial assets
|
3,573
|
3,573
|
4,390
|
4,390
|
||||||||||||
|
Embedded derivative asset
|
-
|
-
|
266
|
266
|
||||||||||||
|
Cash and cash equivalents
|
129,716
|
129,716
|
73,966
|
73,966
|
||||||||||||
|
Total financial assets at amortized cost
|
136,307
|
136,307
|
80,545
|
80,545
|
||||||||||||
|
Fair value of financial liabilities
|
2020
|
2019
|
||||||||||||||
|
Carrying
|
Carrying
|
|||||||||||||||
|
amount
|
Fair value
|
amount
|
Fair value
|
|||||||||||||
|
£
|
’000
|
£
|
’000
|
£
|
’000
|
£
|
’000
|
|||||||||
|
Financial liabilities at amortized cost
|
||||||||||||||||
|
Trade payables
|
25,084
|
25,084
|
15,579
|
15,579
|
||||||||||||
|
Interest-bearing loans and borrowings (Notes 18 and 21)
|
36,654
|
36,654
|
19,157
|
19,157
|
||||||||||||
|
Derivative liability
|
-
|
-
|
5,127
|
5,127
|
||||||||||||
|
Total financial liabilities
|
61,738
|
61,738
|
39,863
|
39,863
|
||||||||||||
| 26. |
Financial instruments (continued)
|
| 26. |
Financial instruments (continued)
|
|
At
January
1, 2020
£’000
|
Cash
flows
£’000
|
Foreign
exchange
movement
£’000
|
Net
finance
(income) /
costs
£’000
|
Leases
£’000
|
Other
£’000
|
At
December
31, 2020
£’000
|
||||||||||||||||||||||
|
Interest-bearing loans and borrowings
|
19,157
|
37,252
|
(1,306
|
)
|
708
|
(19,157
|
)
|
36,654
|
||||||||||||||||||||
|
Derivative liability
|
5,127
|
-
|
-
|
(1,287
|
)
|
-
|
(3,840
|
)
|
-
|
|||||||||||||||||||
|
Lease liabilities
|
40,250
|
(4,426
|
)
|
-
|
(8,591
|
)
|
27,233
|
|||||||||||||||||||||
|
Total liabilities from financing activities
|
64,534
|
32,826
|
(1,306
|
)
|
(579
|
)
|
(8,591
|
)
|
(22,997
|
)
|
63,887
|
|||||||||||||||||
|
At
January
1, 2019
£’000
|
Cash
flows
£’000
|
Foreign
exchange
movement
£’000
|
Net finance
(income) / costs
£’000
|
Leases
£’000
|
At
December
31, 2019
£’000
|
|||||||||||||||||||
|
Interest-bearing loans and borrowings
|
18,878
|
-
|
(563
|
)
|
842
|
-
|
19,157
|
|||||||||||||||||
|
Derivative liability
|
-
|
-
|
-
|
5,127
|
-
|
5,127
|
||||||||||||||||||
|
Lease liabilities
|
46,555
|
(4,036
|
)
|
9
|
2,938
|
(5,216
|
)
|
40,250
|
||||||||||||||||
|
Total liabilities from financing activities
|
65,433
|
(4,036
|
)
|
(554
|
)
|
8,907
|
(5,216
|
)
|
64,534
|
|||||||||||||||
| 26. |
Financial instruments (continued)
|
| 27. |
Post-employment benefit plans
|
| 28. |
Commitments and contingencies
|
|
As at December 31, 2020
|
Less than 1
year
|
1-3
years
|
3-5
years
|
More than 5
years
|
Total
|
|||||||||||||||
|
Lease liabilities – existing
|
3,529
|
5,322
|
4,286
|
32,600
|
45,737
|
|||||||||||||||
|
Lease liabilities – contingent
|
-
|
2,254
|
2,471
|
1,841
|
6,566
|
|||||||||||||||
|
Manufacturing
|
2,824
|
500
|
-
|
-
|
3,324
|
|||||||||||||||
|
Capital commitments
|
77
|
-
|
-
|
-
|
77
|
|||||||||||||||
|
Total contractual obligations
|
6,430
|
8,076
|
6,757
|
34,441
|
55,704
|
|||||||||||||||
|
As at December 31, 2019
|
Less than 1
year
|
1-3
years
|
3-5
years
|
More than 5
years
|
Total
|
|||||||||||||||
|
Lease liabilities – existing
|
4,469
|
8,958
|
7,876
|
45,288
|
66,591
|
|||||||||||||||
|
Lease liabilities - contingent
|
68 |
1,604 |
2,685 |
2,688 |
7,045 |
|||||||||||||||
|
Manufacturing
|
3,669
|
642
|
-
|
-
|
4,311
|
|||||||||||||||
|
Capital commitments
|
1,460
|
-
|
-
|
-
|
1,460
|
|||||||||||||||
|
Total contractual obligations
|
9,666
|
11,204
|
10,561
|
47,976
|
79,407
|
|||||||||||||||
| 29. |
Related party disclosures
|
|
2020
|
2019
|
2018
|
||||||||||||||||||||||
|
Sales to
related
party
£000’s
|
Purchases
from related
party
£000’s
|
Sales to
related
party
£000’s
|
Purchases
from related
party
£000’s
|
Sales to
related
party
£000’s
|
Purchases
from related
party
£000’s
|
|||||||||||||||||||
|
Aigenpulse Limited
|
-
|
-
|
-
|
500
|
-
|
729
|
||||||||||||||||||
|
Adaptimmune Limited
|
-
|
-
|
-
|
-
|
69
|
-
|
||||||||||||||||||
|
Malin Life Sciences Holdings Limited
|
-
|
-
|
-
|
-
|
-
|
2
|
||||||||||||||||||
|
Oxford Nanosystems Limited
|
-
|
-
|
-
|
-
|
2
|
-
|
||||||||||||||||||
|
Oxford Innovation Ltd
|
-
|
-
|
-
|
30
|
-
|
13
|
||||||||||||||||||
|
-
|
-
|
-
|
530
|
71
|
744
|
|||||||||||||||||||
|
2020
£000’s
|
2019
£000’s
|
2018
£’000’s
|
||||||||||
|
Short-term employee benefits
|
3,421
|
6,502
|
4,435
|
|||||||||
|
Share-based payments
|
5,602
|
3,667
|
270
|
|||||||||
|
9,023
|
10,169
|
4,705
|
||||||||||
|
30.
|
Events after the reporting period
|
| 30. |
Events after the reporting period (continued)
|
Exhibit 1.1
Company number: 13119746
IMMUNOCORE HOLDINGS PLC
ARTICLES OF ASSOCIATION
Adopted by special resolution on 3 February 2021

Cooley (UK) LLP, Dashwood, 69 Old Broad Street, London EC2M 1QS, UK
T: +44 (0) 20 7583 4055 F: +44 (0) 20 7785 9355 www.cooley.com
Contents
Clause
| 1. Exclusion of model articles (and any other prescribed regulations) | 1 |
| 2. Interpretation | 1 |
| 3. Form of resolution | 5 |
| 4. Limited liability | 5 |
| 5. Change of name | 5 |
| 6. Shareholder rights | 5 |
| 7. Power to attach rights to shares | 9 |
| 8. Allotment of shares and pre-emption | 9 |
| 9. Redeemable shares | 11 |
| 10. Pari passu issues | 11 |
| 11. Variation of rights | 11 |
| 12. Rights deemed not varied | 12 |
| 13. Payment of commission | 12 |
| 14. Trusts not recognised | 12 |
| 15. Uncertificated shares | 12 |
| 16. Share certificates | 13 |
| 17. Replacement certificates | 14 |
| 18. Lien on shares not fully paid | 15 |
| 19. Enforcement of lien by sale | 15 |
| 20. Application of proceeds of sale | 15 |
| 21. Calls | 15 |
| 22. Liability of joint holders | 16 |
| 23. Interest on calls | 16 |
| 24. Sums treated as calls | 16 |
| 25. Power to differentiate | 16 |
| 26. Payment of calls in advance | 16 |
| 27. Notice if call or instalment not paid | 17 |
| 28. Forfeiture for non-compliance | 17 |
| 29. Notice after forfeiture | 17 |
| 30. Forfeiture may be annulled | 17 |
| 31. Surrender | 17 |
| 32. Sale of forfeited shares | 17 |
| 33. Effect of forfeiture | 18 |
| 34. Evidence of forfeiture | 18 |
| 35. Form of transfer | 18 |
| 36. Right to refuse registration of transfer | 19 |
| 37. Notice of refusal to register a transfer | 19 |
| 38. No fees on registration | 19 |
| 39. Other powers in relation to transfers | 20 |
| 40. Transmission of shares on death | 20 |
| 41. Election of person entitled by transmission | 20 |
| 42. Rights on transmission | 20 |
| 43. Destruction of documents | 21 |
| 44. Sub-division | 22 |
| 45. Fractions | 22 |
| 46. Annual general meetings | 23 |
| 47. Convening and format of general meetings | 23 |
| 48. Notice of general meetings | 25 |
| 49. Contents of notice of general meetings | 25 |
| 50. Omission to give notice and non-receipt of notice | 26 |
| 51. Postponement of general meeting | 26 |
| 52. Quorum at general meeting | 27 |
| 53. Procedure if quorum not present | 27 |
| 54. Chair of general meeting | 27 |
| 55. Entitlement to attend, speak and participate | 27 |
| 56. Adjournments | 28 |
| 57. Notice of adjournment | 29 |
| 58. Business of adjourned meeting | 29 |
| 59. Security arrangements and orderly conduct | 29 |
| 60. Overflow meeting rooms | 30 |
| 61. Amendment to resolutions | 30 |
| 62. Withdrawal and ruling amendments out of order | 31 |
| 63. Members’ resolutions | 31 |
| 64. Method of voting | 31 |
| 65. Objection to error in voting | 31 |
| 66. Voting Procedure | 31 |
| 67. Votes of members | 32 |
| 68. No right to vote where sums overdue on shares | 33 |
| 69. Voting by Proxy | 33 |
| 70. Receipt of proxy | 34 |
| 71. Revocation of proxy | 35 |
| 72. Availability of appointments of proxy | 35 |
| 73. Corporate representatives | 35 |
| 74. Failure to disclose interests in shares | 36 |
| 75. Power of sale of shares of untraced members | 39 |
| 76. Application of proceeds of sale of shares of untraced members | 40 |
| 77. Number of Directors | 40 |
| 78. Power of company to appoint Directors | 40 |
| 79. Power of Board to appoint Directors | 40 |
| 80. Eligibility of new Directors | 41 |
| 81. Classes and Retirement of Directors | 41 |
| 82. Deemed re-appointment | 42 |
| 83. Procedure if insufficient Directors appointed | 42 |
| 84. Removal of Directors | 42 |
| 85. Vacation of office by Director | 43 |
| 86. Resolution as to vacancy conclusive | 43 |
| 87. Appointment of alternate Directors | 44 |
| 88. Alternate Directors’ participation in Board meetings | 44 |
| 89. Alternate Director responsible for own acts | 44 |
| 90. Interests of alternate Director | 44 |
| 91. Revocation of alternate Director | 44 |
| 92. Directors’ fees | 45 |
| 93. Expenses | 45 |
| 94. Additional remuneration | 45 |
| 95. Remuneration of executive Directors | 45 |
| 96. Pensions and other benefits | 46 |
| 97. Powers of the Board | 46 |
| 98. Powers of Directors if less than minimum number | 46 |
| 99. Powers of executive Directors | 47 |
| 100. Delegation to committees | 47 |
| 101. Local management | 47 |
| 102. Power of attorney | 48 |
| 103. Exercise of voting power | 48 |
| 104. Provision for employees on cessation of business | 48 |
| 105. Overseas registers | 48 |
| 106. Borrowing powers | 48 |
| 107. Board meetings | 49 |
| 108. Notice of Board meetings | 49 |
| 109. Quorum | 49 |
| 110. Chair | 49 |
| 111. Voting | 50 |
| 112. Participation by telephone or other form of communication | 50 |
| 113. Resolution in writing | 50 |
| 114. Proceedings of committees | 50 |
| 115. Minutes of proceedings | 50 |
| 116. Validity of proceedings | 51 |
| 117. Transactions or other arrangements with the company | 51 |
| 118. Authorisation of Directors’ conflicts of interest | 51 |
| 119. Directors’ permitted interests | 53 |
| 120. General | 55 |
| 121. Power to authenticate documents | 55 |
| 122. Use of seals | 55 |
| 123. Declaration of dividends | 56 |
| 124. Interim dividends | 56 |
| 125. Calculation and currency of dividends | 56 |
| 126. Amounts due on shares can be deducted from dividends | 57 |
| 127. Dividends not in cash | 57 |
| 128. No interest on dividends | 57 |
| 129. Method of payment | 57 |
| 130. Uncashed dividends | 59 |
| 131. Unclaimed dividends | 59 |
| 132. Scrip dividends | 59 |
| 133. Capitalisation of reserves | 61 |
| 134. Record dates | 63 |
| 135. Inspection of records | 64 |
| 136. Account to be sent to members | 64 |
| 137. Service of Notices | 64 |
| 138. Hard copy form | 66 |
| 139. Electronic form | 66 |
| 140. Electronic means | 66 |
| 141. Website | 67 |
| 142. Sending or supplying any document, information or notice by any other means | 67 |
| 143. Presence at meeting evidence in itself of receipt of notice | 67 |
| 144. Notice on person entitled by transmission | 67 |
| 145. Record date for service | 68 |
| 146. Evidence of service | 68 |
| 147. Notice when post not available | 69 |
| 148. Validation of documents in electronic form | 69 |
| 149. Winding up | 69 |
| 150. Indemnity and insurance | 70 |
| 151. Exclusive jurisdiction | 71 |
THE COMPANIES ACT 2006
PUBLIC COMPANY LIMITED BY SHARES
ARTICLES OF ASSOCIATION
OF
IMMUNOCORE HOLDINGS PLC
(Adopted by special resolution on 3 February 2021)
| 1. | Exclusion of model articles (and any other prescribed regulations) |
No regulations or articles set out in any statute, or in any statutory instrument or other subordinate legislation made under any statute, concerning companies (including the regulations in the Companies (Model Articles) Regulations 2008 (SI 2008/3229)) shall apply as the articles of the Company. The following shall be the articles of association of the Company.
| 2. | Interpretation |
| 2.1 | In these Articles, unless the context otherwise requires: |
Act: the Companies Act 2006;
address: includes any number or address used for the purposes of sending or receiving documents or information by electronic means;
Articles: these articles of association as altered from time to time and “Article” shall be construed accordingly;
Beneficial Ownership Limitation: 9.99% of any class of securities of the Company registered under the Exchange Act, which percentage may be increased or decreased on a holder-by-holder basis by a holder of Non-Voting Ordinary Shares to such other percentage as such holder may designate in writing (with any decrease to be effective upon at least sixty one days’ notice) to the Company, provided, however, that: (i) any such increase shall not exceed 19.9% of any class of securities of the Company registered under the Exchange Act; and (ii) any such increase or decrease shall only be applicable to such holder in relation to such securities. For the purpose of calculating the Beneficial Ownership Limitation, a holder may rely on the number of outstanding shares of the subject class as stated in the most recent of the following: (A) the Company’s most recent periodic or annual filing; (B) a more recent public announcement by the Company that is publicly filed; or (C) a more recent notice by the Company or the Company’s registrar to the holder setting forth the number of shares then outstanding. Upon the written request of a holder (which may be by email with confirmation), the Company shall, within five business days thereof, confirm in writing to such holder (which may be via email) the number of shares then outstanding;
Board: the board of Directors for the time being of the Company or the Directors present or deemed to be present at a duly convened quorate meeting of the Directors;
business day: means any day other than Saturday, Sunday or other day on which commercial banks in New York and/or London are authorised or required by law to remain closed;
certificated shares: a share which is not an uncertificated share and references in these Articles to a share being held in certificated form shall be construed accordingly;
class meeting: has the meaning given to it in Article 11;
clear days: in relation to a period of notice means that period excluding the day when the notice is served or deemed to be served and the day for which it is given or on which it is to take effect;
Companies Acts: the Companies Acts as defined by section 2 of the Act, and includes the uncertificated securities rules and, where the context requires, every other statute (including orders, regulations or other subordinate legislation made under them) from time to time in force concerning companies and affecting the Company;
Company: Immunocore Holdings plc;
default shares: has the meaning given to it in Article 74.1;
Deferred Shares: the Company’s deferred shares with a nominal value of £0.0001 each as sub-divided or consolidated from time to time;
Depositary: the holder of a share for the time being held on behalf of another person on the terms of a depositary agreement or a depositary receipt or a similar document;
Director: a director for the time being of the Company;
elected shares: has the meaning given to it in Article 132.1(j);
electronic form: has the meaning given to it in section 1168 of the Act;
electronic general meeting: has the meaning given to it in Article 47.5;
electronic means: has the meaning given to it in section 1168 of the Act;
Exchange Act: collectively, the U.S. Securities Exchange Act of 1934 and the rules and regulations promulgated thereunder;
FSMA: the Financial Services and Markets Act 2000;
hard copy form: has the meaning given to it in section 1168 of the Act;
hybrid general meeting: has the meaning given to it in Article 47.4;
Interested Director: has the meaning given to it in Article 118.1;
Listing: listing of the Company’s Ordinary Shares (including Ordinary Shares represented by American Depositary Shares) on Nasdaq;
member: a member of the Company, or where the context requires, a member of the Board or of any committee;
Nasdaq: the Nasdaq Stock Market LLC;
Non-Voting Ordinary Shares: the Company’s non-voting ordinary shares with a nominal value of £0.002 each as sub-divided or consolidated from time to time;
Non-Voting Ordinary Share Re-Designation Notice: has the meaning given to it in Article 6.8;
Office: the registered office from time to time of the Company;
Operator: Euroclear UK and Ireland Limited or such other person as may for the time being be approved by HM Treasury as Operator under the uncertificated securities rules;
Ordinary Shares: the Company’s ordinary shares with a nominal value of £0.002 each as sub-divided or consolidated from time to time;
paid up: paid up or credited as paid up;
participating class: a class of shares title to which is permitted by the Operator to be transferred by means of a relevant system;
principal place: has the meaning given to it in Article 47.3;
proxy notification address: has the meaning given to it in Article 70.1(a);
proxy notification electronic address: has the meaning given in Article 70.1(b);
record date: has the meaning given to it in Article 134.1;
Register: the register of members of the Company to be maintained under the Act or as the case may be any overseas branch register maintained under Article 105;
Relevant Interest: has the meaning given to it in Article 119.4;
relevant system: a computer-based system which allows units of securities without written instruments to be transferred and endorsed pursuant to the uncertificated securities rules or other applicable regulations;
Retiring Directors: has the meaning given to it in Article 83.1;
satellite place: has the meaning given to it in Article 47.3;
Seal: the common seal of the Company or, where the context allows, any official seal kept by the Company under section 50 of the Act;
SEC: the United States Securities and Exchange Commission;
Secretary: the secretary of Company for the time being;
section 793 notice: has the meaning given to it in Article 74.1;
Securities Act: the U.S. Securities Act of 1933 and the rules and regulations promulgated thereunder;
share: an Ordinary Share (including, where the context so requires (and save as set out in Article 6.7), a Non-Voting Ordinary Share);
Share Warrant: a warrant to bearer issued by the Company in respect of its shares;
uncertificated securities rules: any provision of the Companies Acts relating to the holding, evidencing of title to, or transfer of uncertificated shares and any legislation, rules or other arrangements made under or by virtue of such provision (including the Uncertificated Securities Regulations 2001 (SI 2001/3755) as amended or replaced from time to time and any subordinate legislation or rules made under them for the time being in force); and
uncertificated share: a share of a class which is at the relevant time a participating class, title to which is recorded on the Register as being held in uncertificated form and references in these Articles to a share being held in uncertificated form shall be construed accordingly.
| 2.2 | Headings are used for convenience only and shall not affect the construction or interpretation of these Articles. |
| 2.3 | Unless the context otherwise requires, a person includes a natural person, corporate or unincorporated body (whether or not having separate legal personality). |
| 2.4 | Unless the context otherwise requires, words in the singular shall include the plural and vice versa. |
| 2.5 | A reference to one gender shall include a reference to the other genders. |
| 2.6 | A reference to a statute or statutory provision is a reference to it as it is in force for the time being, taking account of any amendment, extension, or re-enactment, and includes any subordinate legislation for the time being in force made under it. |
| 2.7 | Any words or expressions defined in the Companies Acts in force when these Articles or any part of these Articles are adopted shall (if not inconsistent with the subject or context in which they appear) have the same meaning in these Articles or that part, save that the word “company” shall include any body corporate. |
| 2.8 | A reference to a document being signed or to signature includes references to its being executed under hand or under seal or by any other method and, in the case of a communication in electronic form, such references are to its being authenticated as specified by the Companies Acts. |
| 2.9 | A reference to writing or written includes references to any method of representing or reproducing words in a legible and non-transitory form whether sent or supplied in electronic form or otherwise. |
| 2.10 | A reference to documents or information being sent or supplied by or to a company (including the Company) shall be construed in accordance with section 1148(3) of the Act. |
| 2.11 | A reference to a meeting: |
| (a) | shall mean a meeting convened and held in any manner permitted by these Articles, including without limitation a general meeting at which some or all of those persons entitled to be present attend and participate by means of electronic facility or facilities, and such persons shall be deemed to be present at that meeting for all purposes of the Companies Acts and these Articles, and present, attend, participate, being present, attending, participating, presence, attendance and participation shall be construed accordingly; and |
| (b) | shall not be taken as requiring more than one person to be present if any quorum requirement can be satisfied by one person. |
| 2.12 | References to electronic facility mean a device, system, procedure, method or facility providing an electronic means of attendance at or participation in (or both attendance at and participation in) a general meeting as determined by the Directors pursuant to Article 47.4 or 47.5, including without limitation online platforms, application technology and conference call systems. |
| 3. | Form of resolution |
Subject to the Companies Acts, where anything can be done by passing an ordinary resolution, this can also be done by passing a special resolution.
| 4. | Limited liability |
The liability of the members of the Company is limited to the amount, if any, unpaid on the shares in the Company held by them.
| 5. | Change of name |
The Company may change its name by resolution of the Board.
| 6. | Shareholder rights |
| 6.1 | The Ordinary Shares shall rank pari passu as a single class. The Deferred Shares shall rank pari passu as a single class. The Non-Voting Ordinary Shares shall rank pari passu as a single class and pari passu with the Ordinary Shares save as set out in Article 6.7 below. |
| 6.2 | In the event of the liquidation, dissolution or winding up of the Company, the assets of the Company available for distribution to members shall be distributed amongst all holders of the Ordinary Shares and Non-Voting Ordinary Shares (if any) in proportion to the number of shares held irrespective of the amount paid or credited as paid on any share. |
| 6.3 | Any: |
| (a) | consolidation or merger of the Company with or into another entity or entities (whether or not the Company is the surviving entity) as a result of which the holders of the Company’s outstanding shares possessing the voting power (under ordinary circumstances) to elect a majority of the Board immediately prior to such sale or issue cease to own the Company’s outstanding shares possessing the voting power (under ordinary circumstances) to elect a majority of the Board; |
| (b) | sale or transfer by the Company of all or substantially all of its assets (determined either for the Company alone or together with its subsidiaries on a consolidated basis); or |
| (c) | sale, transfer or issuance or series of sales, transfers and/or issues of shares by the Company or the holders thereof, as a result of which the holders of the Company’s outstanding shares possessing the voting power (under ordinary circumstances) to elect a majority of the Board immediately prior to such sale or issue cease to own the Company’s outstanding shares possessing the voting power (under ordinary circumstances) to elect a majority of the Board, |
shall be deemed to be a liquidation, dissolution and winding up of the Company for purposes of Article 6.2 (unless the Board determine otherwise), and the holders of the Ordinary Shares and Non-Voting Ordinary Shares (if any) shall be entitled to receive from the Company the amounts payable with respect to the Ordinary Shares and Non-Voting Ordinary Shares (if any) on a liquidation, dissolution or winding up of the Company under Article 6.2 in cancellation of their Ordinary Shares or Non-Voting Ordinary Shares (if any) upon the completion of any such transaction.
| 6.4 | At a general meeting of the Company and at any separate class meeting of the holders of Ordinary Shares, where a holder of Ordinary Shares is entitled to vote, such holder is entitled to one vote for each Ordinary Share held. |
| 6.5 | A holder of Ordinary Shares is entitled to receive notice of any general meeting of the Company (and notice of any separate class meeting of the holders of Ordinary Shares) and a copy of every report, accounts, circular or other document sent out by the Company to members. |
| 6.6 | Notwithstanding any other provision of these Articles, the special rights, privileges, restrictions and limitations attaching to the Deferred Shares are as follows: |
| (a) | the Deferred Shares shall not be entitled to any dividends or to any other right of participation in the income or profits of the Company; |
| (b) | on the return of assets on a winding-up of the Company, the Deferred Shares shall confer on the holders thereof an entitlement to receive out of the assets of the Company available for distribution amongst the members (subject to the rights of any new class of shares with preferred rights) the amount paid up or credited as paid up on the Deferred Shares held by them respectively after (but only after) payment shall have been made to the holders of the Ordinary Shares and Non-Voting Ordinary Shares (if any) of the amounts paid up or credited as paid up on such shares and the sum of £1,000,000 in respect of each Ordinary Share or Non-Voting Ordinary Share held by them respectively. The Deferred Shares shall confer on the holders thereof no further right to participate in the assets of the Company; |
| (c) | the Deferred Shares do not entitle the holder thereof to receive notice of or to attend, speak or vote at any general meeting of the Company, or be part of the quorum thereof as the holders of the Deferred Shares; |
| (d) | any reduction of capital involving the cancellation of the Deferred Shares for no consideration shall not be deemed to be a variation of the rights attaching to them nor a modification or abrogation of the rights or privileges attaching to the Deferred Shares; |
| (e) | the special rights conferred upon the holders of the Deferred Shares shall be deemed not to be modified, varied or abrogated by the creation or issue of further shares ranking pari passu with or in priority to the Deferred Shares; |
| (f) | the Deferred Shares shall not entitle the holder to receive a share certificate in respect of such shareholding, save as required by law; |
| (g) | no transfer of any Deferred Shares shall be permitted save as provided in Article 6.6(h); |
| (h) | the Company shall have irrecoverable authority from each holder of the Deferred Shares at any time to do all or any of the following without obtaining the sanction of the holder or holders of the Deferred Shares: |
| (i) | to appoint any person to execute on behalf of any holder of Deferred Shares a transfer of all or any of those shares and/or an agreement to transfer the same (without making any payment for them) to such person or persons as the Company may determine and to execute any other documents which such person may consider necessary or desirable to effect such transfer, in each case without obtaining the sanction of the holder(s) and without any payment being made in respect of such acquisition; and |
| (ii) | to purchase all or any of the Deferred Shares without obtaining the consent of the holders of those shares in consideration for an amount not exceeding £1.00 in respect of all the Deferred Shares then being purchased and: |
| (A) | for the purpose of any such purchase, to appoint any person to execute an instrument of transfer in respect of such shares to the Company on behalf of any holder of Deferred Shares; and |
| (B) | to cancel all or any of the Deferred Shares purchased. |
| 6.7 | The Non-Voting Ordinary Shares shall have the same rights and restrictions as the Ordinary Shares and shall otherwise rank pari passu in all respects with the Ordinary Shares and a holder of Non-Voting Ordinary Shares shall be subject to the same obligations and liabilities as a holder of Ordinary Shares save as set out below: |
| (a) | a holder of Non-Voting Ordinary Shares shall, in relation to the Non-Voting Ordinary Shares held by him or her, have no right to receive notice of, or to attend or vote at, any general meeting of shareholders save in relation to a variation of class rights of the Non-Voting Ordinary Shares. At any such general meeting of the Company in relation to a variation of class rights of the Non-Voting Ordinary Shares and at any separate class meeting of the holders of Non-Voting Ordinary Shares, where a holder of Non-Voting Ordinary Shares is entitled to vote, such holder is entitled to one vote for each Non-Voting Ordinary Share held; and |
| (b) | the Non-Voting Ordinary Shares shall be re-designated as Ordinary Shares by the Company (acting by the Board, or a duly authorised committee or representative thereof): |
| (i) | upon delivery by a holder of Non-Voting Ordinary Shares to the Company of a Non-Voting Ordinary Share Re-Designation Notice (as defined in Article 6.8 below) and otherwise subject to the terms and conditions set out in Article 6.8; and/or |
| (ii) | automatically upon a transfer of a Non-Voting Ordinary Share by its holder to any person that is not an “affiliate” or “group member” with whom such holder is required to aggregate beneficial ownership for purposes of section 13(d) of the Exchange Act. For the avoidance of doubt, the automatic re-designation under this Article 6.7(b)(ii) shall only be in respect of the Non-Voting Ordinary Share(s) that is/are the subject of such transfer and not any other Non-Voting Ordinary Shares held by the holder. |
| 6.8 | A holder of Non-Voting Ordinary Shares may elect to have some or all of their Non-Voting Ordinary Shares re-designated as Ordinary Shares by providing a written notice (a “Non-Voting Ordinary Share Re-Designation Notice”) to the Company, specifying the number of Non-Voting Ordinary Shares it wishes to have re-designated as Ordinary Shares and including instructions as to whether the relevant Ordinary Shares are to be held in certificated or uncertificated form in accordance with Article 6.10(c) and in the case of Ordinary Shares to be held in uncertificated form the details of the relevant account of the holder of Non-Voting Ordinary Shares’ broker into which they are to be credited in accordance with Article 6.10(c)(ii), and being accompanied by the relevant share certificate(s) (or indemnity in respect of such share certificate or other evidence as the Company may require) in respect of the relevant Non-Voting Ordinary Shares, save that a holder of Non-Voting Ordinary Shares shall not be entitled to have any Non-Voting Ordinary Shares re-designated as Ordinary Shares where such re-designation would result in such holder thereof beneficially owning (for purposes of section 13(d) of the Exchange Act), when aggregated with “affiliates” and “group” members with whom such holder is required to aggregate beneficial ownership for purposes of section 13(d) of the Exchange Act, in excess of the Beneficial Ownership Limitation (and the Company shall be entitled to receive written confirmation from such holder of this fact prior to the re-designation as Ordinary Shares of the relevant Non-Voting Ordinary Shares). |
| 6.9 | Within three business days following delivery of a Non-Voting Ordinary Share Re-Designation Notice to the Company, and such documentation and/or confirmations as the Company may reasonably request as specifically provided for in Article 6.8, the relevant Non-Voting Ordinary Shares shall be re-designated as Ordinary Shares by the Board, or a duly authorised committee or representative thereof. |
| 6.10 | Following any re-designation of Non-Voting Ordinary Shares in accordance with Article 6.7(b)(i), the Company shall: |
| (a) | procure that the Register is updated to reflect the re-designation; |
| (b) | where less than all of the Non-Voting Ordinary Shares represented by any certificate delivered in accordance with Article 6.8 are re-designated as Ordinary Shares, issue and deliver to the holder a new certificate in respect of the balance of Non-Voting Ordinary Shares comprised in the surrendered certificate within fourteen days of the date of re-designation to such holder, by post to its address as shown in the Register, at his, her or its own risk and free of charge; and |
| (c) | either: |
| (i) | where the Ordinary Shares into which the Non-Voting Ordinary Shares are to be re-designated are to be held in certificated form, issue and deliver to the holder a new certificate in respect of the appropriate number of Ordinary Shares within fourteen days of the date of re-designation to such holder, by post to its address as shown in the Register, at his, her or its own risk and free of charge; or |
| (ii) | where the Ordinary Shares into which the Non-Voting Ordinary Shares are to be re-designated are to be held in uncertificated form, procure that the appropriate number of Ordinary Shares are credited to the relevant account of the holder of Non-Voting Ordinary Shares’ broker in the relevant system as specified in the Non-Voting Ordinary Share Re-Designation Notice within two business days of the date of re-designation. |
| 6.11 | Upon the re-designation of the Non-Voting Ordinary Shares as Ordinary Shares, such Ordinary Shares shall rank pari passu with the other Ordinary Shares of the Company in all respects. |
| 7. | Power to attach rights to shares |
Subject to the Companies Acts and to any rights attached to existing shares, any share may be issued with or have attached to it such rights and restrictions as the Company may by ordinary resolution determine, or if no ordinary resolution has been passed or so far as the resolution does not make specific provision, as the Board may determine.
| 8. | Allotment of shares and pre-emption |
| 8.1 | Subject to the Companies Acts, these Articles and to any relevant authority of the Company in general meeting required by the Act, the Board may offer, allot (with or without conferring rights of renunciation), grant options over or otherwise deal with or dispose of shares or grant rights to subscribe for or convert any security into shares to such persons, at such times and upon such terms as the Board may decide. No share may be issued at a discount to the nominal value of such share. |
| 8.2 | The Board may, at any time after the allotment of any share but before any person has been entered in the Register, recognise a renunciation by the allottee in favour of some other person and accord to the allottee of a share a right to effect such renunciation and/or allow the rights to be represented to be one or more participating securities, in each case upon and subject to such terms and conditions as the Board may think fit to impose. |
| 8.3 | Under and in accordance with section 551 of the Act, the Directors shall be generally and unconditionally authorised to exercise for each prescribed period all the powers of the Company to allot shares and to grant rights to subscribe for, or to convert any security into, shares up to an aggregate nominal amount equal to the Section 551 Amount. |
| 8.4 | Under and within the terms of the said authority or otherwise in accordance with section 570 of the Act, the Directors shall be empowered during each prescribed period to allot equity securities (as defined by the Act) wholly for cash: |
| (a) | in connection with a rights issue; and |
| (b) | otherwise than in connection with a rights issue up to an aggregate nominal amount equal to the Section 561 Amount. |
| 8.5 | During each prescribed period the Company and its Directors by such authority and power may make offers or agreements which would or might require equity securities or other securities to be allotted after the expiry of such period. |
| 8.6 | For the purposes of this Article 8: |
| (a) | rights issue means an offer of equity securities (as defined by the Act) open for acceptance for a period fixed by the Board to holders of equity securities on the Register on a fixed record date in proportion to their respective holdings of such securities or in accordance with the rights attached to them but subject to such exclusions or other arrangements as the Board may deem necessary or expedient with regard to treasury shares, fractional entitlements or legal or practical problems under the laws of any territory or under the requirements of any recognised regulatory body or stock exchange in any territory; |
| (b) | prescribed period means any period (not exceeding five years on any occasion) for which the authority, in the case of Article 8.3, is conferred or renewed by ordinary or special resolution stating the Section 551 Amount and in the case of Article 8.4 is conferred or renewed by special resolution stating the Section 561 Amount; |
| (c) | Section 551 Amount means for any prescribed period, the amount stated in the relevant ordinary or special resolution; |
| (d) | Section 561 Amount means for any prescribed period, the amount stated in the relevant special resolution; and |
| (e) | the nominal amount of any securities shall be taken to be, in the case of rights to subscribe for or to convert any securities into shares of the Company, the nominal amount of such shares which may be allotted pursuant to such rights. |
| 9. | Redeemable shares |
Subject to the Companies Acts and to any rights attaching to existing shares, any share may be issued which can be redeemed or is liable to be redeemed at the option of the Company or the holder. The Board may determine the terms, conditions and manner of redemption of any redeemable shares which are issued. Such terms and conditions shall apply to the relevant shares as if the same were set out in these Articles.
| 10. | Pari passu issues |
If new shares are created or issued which rank equally with any other existing shares, or the Company purchases any of its own shares, the rights of the existing shares will not be regarded as changed or abrogated unless the terms of the existing shares expressly say otherwise.
| 11. | Variation of rights |
| 11.1 | Subject to the Companies Acts, the rights attached to any class of shares can be varied or abrogated: |
| (a) | in such manner (if any) as may be provided by those rights; |
| (b) | with the consent in writing of the holders of not less than three-quarters in nominal value of the issued shares of that class (excluding any shares of that class held as treasury shares); or |
| (c) | with the authority of a special resolution passed at a separate meeting of the holders of the relevant class of shares known as a class meeting. |
| 11.2 | The provisions of this Article will apply to any variation or abrogation of rights of shares forming part of a class. Each part of the class which is being treated differently is treated as a separate class in applying this Article. |
| 11.3 | All the provisions in these Articles as to general meetings shall apply, with any necessary modifications, to every class meeting except that: |
| (a) | the quorum at every such meeting shall not be less than two persons holding or representing by proxy at least one-third of the nominal amount paid up on the issued shares of the class (excluding any shares of that class held as treasury shares); and |
| (b) | if at any adjourned meeting of such holders such quorum as set out above is not present, at least one person holding shares of the class who is present in person or by proxy shall be a quorum. |
| 11.4 | The Board may convene a class meeting whenever it thinks fit and whether or not the business to be transacted involves a variation or abrogation of class rights. |
| 12. | Rights deemed not varied |
Unless otherwise expressly provided by the rights attached to any class of shares, those rights shall be deemed not to be varied by the purchase by the Company of any of its own shares or the holding of such shares as treasury shares.
| 13. | Payment of commission |
The Company may in connection with the issue of any shares or the sale for cash of treasury shares exercise all powers of paying commission and brokerage conferred or permitted by the Companies Acts. Any such commission or brokerage may be satisfied by the payment of cash or by the allotment of fully or partly paid shares or other securities or the grant of an option to call for an allotment of shares or any combination of such methods.
| 14. | Trusts not recognised |
Except as otherwise expressly provided by these Articles, required by law or as ordered by a court of competent jurisdiction, the Company shall not recognise any person as holding any share on any trust, and the Company shall not be bound by or required in any way to recognise (even when having notice of it) any equitable, contingent, future, partial or other claim to or interest in any share other than an absolute right of the holder of the whole of the share.
| 15. | Uncertificated shares |
| 15.1 | Under and subject to the uncertificated securities rules, the Board may permit title to shares of any class to be evidenced otherwise than by certificate and title to shares of such a class to be transferred by means of a relevant system and may make arrangements for a class of shares (if all shares of that class are in all respects identical) to become a participating class. Title to shares of a particular class may only be evidenced otherwise than by a certificate where that class of shares is at the relevant time a participating class. The Board may also, subject to compliance with the uncertificated securities rules, determine at any time that title to any class of shares may from a date specified by the Board no longer be evidenced otherwise than by a certificate or that title to such a class shall cease to be transferred by means of any particular relevant system. |
| 15.2 | In relation to a class of shares which is a participating class and for so long as it remains a participating class, no provision of these Articles shall apply or have effect to the extent that it is inconsistent in any respect with: |
| (a) | the holding of shares of that class in uncertificated form; |
| (b) | the transfer of title to shares of that class by means of a relevant system; or |
| (c) | any provision of the uncertificated securities rules, |
and, without prejudice to the generality of this Article, no provision of these Articles shall apply or have effect to the extent that it is in any respect inconsistent with the maintenance, keeping or entering up by the Operator, so long as that is permitted or required by the uncertificated securities rules, of an Operator register of securities in respect of that class of shares in uncertificated form.
| 15.3 | Shares of a class which is at the relevant time a participating class may be changed from uncertificated to certificated form, and from certificated to uncertificated form, in accordance with and subject as provided in the uncertificated securities rules. |
| 15.4 | If, under these Articles or the Companies Acts, the Company is entitled to sell, transfer or otherwise dispose of, forfeit, re-allot, accept the surrender of or otherwise enforce a lien over an uncertificated share, then, subject to these Articles and the Companies Acts, such entitlement shall include the right of the Board to: |
| (a) | require the holder of the uncertificated share by notice in writing to change that share from uncertificated to certificated form within such period as may be specified in the notice and keep it as a certificated share for as long as the Board requires; |
| (b) | appoint any person to take such other steps, by instruction given by means of a relevant system or otherwise, in the name of the holder of such share as may be required to effect the transfer of such share and such steps shall be as effective as if they had been taken by the registered holder of that share; and |
| (c) | take such other action that the Board considers appropriate to achieve the sale, transfer, disposal, forfeiture, re-allotment or surrender of that share or otherwise to enforce a lien in respect of that share. |
| 15.5 | Unless the Board determines otherwise, shares which a member holds in uncertificated form shall be treated as separate holdings from any shares which that member holds in certificated form but a class of shares shall not be treated as two classes simply because some shares of that class are held in certificated form and others in uncertificated form. |
| 15.6 | Unless the Board determines otherwise or the uncertificated securities rules require otherwise, any shares issued or created out of or in respect of any uncertificated shares shall be uncertificated shares and any shares issued or created out of or in respect of any certificated shares shall be certificated shares. |
| 15.7 | The Company shall be entitled to assume that the entries on any record of securities maintained by it in accordance with the uncertificated securities rules and regularly reconciled with the relevant Operator register of securities are a complete and accurate reproduction of the particulars entered in the Operator register of securities and shall accordingly not be liable in respect of any act or thing done or omitted to be done by or on behalf of the Company in reliance on such assumption. Any provision of these Articles which requires or envisages that action will be taken in reliance on information contained in the Register shall be construed to permit that action to be taken in reliance on information contained in any relevant record of securities (as so maintained and reconciled). |
| 16. | Share certificates |
| 16.1 | Other than as provided in Article 6.6(f), every person (except a person to whom the Company is not by law required to issue a certificate) whose name is entered in the Register as a holder of any certificated shares shall be entitled, without charge, to receive within the time limits prescribed by the Companies Acts (unless the terms of issue prescribe otherwise) one certificate for all of the shares of that class registered in his or her name. |
| 16.2 | The Company shall not be bound to issue more than one certificate in respect of shares held jointly by two or more persons. Delivery of a certificate to the person first named in the Register shall be sufficient delivery to all joint holders. |
| 16.3 | Where a member has transferred part only of the shares comprised in a certificate, he or she shall be entitled without charge to a certificate for the balance of such shares to the extent that the balance is to be held in certificated form. Where a member receives more shares of any class, he or she shall be entitled without charge to a certificate for the extra shares of that class to the extent that the balance is to be held in certificated form. |
| 16.4 | A share certificate may be issued under Seal (by affixing the Seal to or printing the Seal or a representation of it on the certificate) or signed by at least two Directors or by at least one Director and the Secretary. Such certificate shall specify the number and class of the shares in respect of which it is issued and the amount or respective amounts paid up on it. The Board may by resolution decide, either generally or in any particular case or cases, that any signatures on any share certificates need not be autographic but may be applied to the certificates by some mechanical or other means or may be printed on them or that the certificates need not be signed by any person. |
| 16.5 | Every share certificate sent in accordance with these Articles will be sent at the risk of the member or other person entitled to the certificate. The Company will not be responsible for any share certificate lost or delayed in the course of delivery. |
| 17. | Replacement certificates |
| 17.1 | Any two or more certificates representing shares of any one class held by any member may at his or her request be cancelled and a single new certificate for such shares issued in lieu without charge on surrender of the original certificates for cancellation. |
| 17.2 | Any certificate representing shares of any one class held by any member may at his or her request be cancelled and two or more certificates for such shares may be issued instead. |
| 17.3 | If a share certificate is defaced, worn out or said to be stolen, lost or destroyed, it may be replaced on such terms as to evidence and indemnity in respect of such share certificate only as the Board may decide and, where it is defaced or worn out, after delivery of the old certificate to the Company. |
| 17.4 | The Board may require the payment of any exceptional out-of-pocket expenses of the Company incurred in connection with the issue of any certificates under this Article. In the case of shares held jointly by several persons, any such request as is mentioned in this Article may be made by any one of the joint holders. |
| 18. | Lien on shares not fully paid |
The Company shall have a first and paramount lien on every share, not being a fully paid share, for all amounts payable to the Company (whether presently or not) in respect of that share. The Company’s lien over a share takes priority over any third party’s interest in that share, and extends to any dividend or other money payable by the Company in respect of that share (and, if the lien is enforced and the share is sold by the Company, the proceeds of sale of that share). The Board may at any time, either generally or in any particular case, waive any lien that has arisen or declare any share to be wholly or in part exempt from the provisions of this Article.
| 19. | Enforcement of lien by sale |
The Company may sell, in such manner as the Board may decide, any share over which the Company has a lien if a sum in respect of which the lien exists is presently payable and is not paid within fourteen clear days after a notice has been served on the holder of the share or the person who is entitled by transmission to the share, demanding payment and stating that if the notice is not complied with the share may be sold. For giving effect to the sale, in the case of a certificated share, the Board may authorise some person to sign an instrument of transfer of the share sold to, or in accordance with the directions, of the buyer. In the case of an uncertificated share, the Board may require the Operator to convert the share into certificated form and after such conversion, authorise any person to sign the instrument of transfer of the share to effect the sale of the share. The buyer shall not be bound to see to the application of the purchase money, nor shall his or her title to the share be affected by any irregularity or invalidity in the proceedings in reference to the sale.
| 20. | Application of proceeds of sale |
The net proceeds of any sale of shares subject to any lien, after payment of the costs, shall be applied:
| (a) | first, in or towards satisfaction of so much of the amount due to the Company or of the liability or engagement (as the case may be) as is presently payable or is liable to be presently fulfilled or discharged; and |
| (b) | second, any residue shall be paid to the person who was entitled to the share at the time of the sale but only after the certificate for the shares sold has been surrendered to the company for cancellation, or an indemnity in a form reasonably satisfactory to the Directors has been given for any lost certificates, and subject to a like lien for debts or liabilities not presently payable as existed on the share prior to the sale. |
| 21. | Calls |
| 21.1 | Subject to these Articles and the terms on which the shares are allotted, the Board may from time to time make calls on the members in respect of any monies unpaid on their shares (whether in respect of nominal value or premium) and not payable on a date fixed by or in accordance with the terms of issue. |
| 21.2 | Each member shall (subject to the Company serving upon him or her at least fourteen clear days’ notice specifying when and where payment is to be made and whether or not by instalments) pay to the Company as required by the notice the amount called on for his or her shares. |
| 21.3 | A call shall be deemed to have been made at the time when the resolution of the Board authorising the call was passed. |
| 21.4 | A call may be revoked or postponed, in whole or in part, as the Board may decide. |
| 21.5 | Liability to pay a call is not extinguished or transferred by transferring the shares in respect of which the call is required to be paid. |
| 22. | Liability of joint holders |
The joint holders of a share shall be jointly and severally liable to pay all calls in respect of the share.
| 23. | Interest on calls |
If a call remains unpaid after it has become due and payable, the person from whom it is due and payable shall pay all expenses that have been incurred by the Company by reason of such non-payment together with interest on the amount unpaid from the day it is due and payable to the time of actual payment at such rate (not exceeding the Bank of England base rate by more than five percentage points) as the Board may decide. The Board may waive payment of the interest or the expenses in whole or in part.
| 24. | Sums treated as calls |
An amount payable in respect of a share on allotment or at any fixed date, whether in respect of nominal value or premium or as an instalment of a call, shall be deemed to be a call and if it is not paid these Articles shall apply as if that sum had become due and payable by virtue of a call.
| 25. | Power to differentiate |
On or before the issue of shares, the Board may decide that allottees or holders of shares can be called on to pay different amounts or that they can be called on at different times.
| 26. | Payment of calls in advance |
The Board may, if it thinks fit, receive from any member willing to advance the same, all or any part of the monies uncalled and unpaid on the shares held by him or her. Such payment in advance of calls shall, to the extent of the payment, extinguish the liability on the shares on which it is made. The Company may pay interest on the money paid in advance, or so much of it as exceeds the amount for the time being called upon the shares in respect of which such advance has been made, at such rate as the Board may decide. The Board may at any time repay the amount so advanced by giving at least three months’ notice in writing to such member of its intention to do so, unless before the expiration of such notice the amount so advanced shall have been called up on the shares in respect of which it was advanced.
| 27. | Notice if call or instalment not paid |
If any member fails to pay the whole of any call (or any instalment of any call) by the date when payment is due, the Board may at any time give notice in writing to such member (or to any person entitled to the shares by transmission), requiring payment of the amount unpaid (and any accrued interest and any expenses incurred by the Company by reason of such non-payment) by a date not less than fourteen clear days from the date of the notice. The notice shall name the place where the payment is to be made and state that, if the notice is not complied with, the shares in respect of which such call was made will be liable to be forfeited.
| 28. | Forfeiture for non-compliance |
If the notice referred to in Article 27 is not complied with, any share for which it was given may be forfeited, by resolution of the Board to that effect, at any time before the payment required by the notice has been made. Such forfeiture shall include all dividends declared or other monies payable in respect of the forfeited shares and not paid before the forfeiture.
| 29. | Notice after forfeiture |
When any share has been forfeited, notice of the forfeiture shall be served on the holder of the share or the person entitled to such share by transmission (as the case may be) before forfeiture. An entry of such notice having been given and of the forfeiture and the date of forfeiture shall immediately be made in the Register in respect of such share. However, no forfeiture shall be invalidated by any omission to give such notice or to make such entry in the Register.
| 30. | Forfeiture may be annulled |
The Board may annul the forfeiture of a share, at any time before any forfeited share has been cancelled or sold, re-allotted or otherwise disposed of, on the terms that payment shall be made of all calls and interest due on it and all expenses incurred in respect of the share and on such further terms (if any) as the Board shall see fit.
| 31. | Surrender |
The Board may accept the surrender of any share liable to be forfeited and, in any event, references in these Articles to forfeiture shall include surrender.
| 32. | Sale of forfeited shares |
| 32.1 | A forfeited share shall become the property of the Company. |
| 32.2 | Subject to the Companies Acts, any such share may be sold, re-allotted or otherwise disposed of, on such terms and in such manner as the Board thinks fit. |
| 32.3 | The Board may, for the purposes of the disposal, authorise some person to transfer the share in question and may enter the name of the transferee in respect of the transferred share in the Register even if no share certificate is lodged and may issue a new certificate to the transferee. An instrument of transfer executed by that person shall be as effective as if it had been executed by the holder of, or the person entitled by transmission to, the share. The Company may receive the consideration (if any) given for the share on its disposal. |
| 33. | Effect of forfeiture |
A member whose shares have been forfeited shall cease to be a member in respect of such forfeited shares and shall surrender the certificate for such shares to the Company for cancellation. Such member shall remain liable to pay to the Company all sums which at the date of forfeiture were presently payable by him or her to the Company in respect of such shares with interest (not exceeding the Bank of England base rate by two percentage points) from the date of the forfeiture to the date of payment. The Directors may waive payment of interest wholly or in part and may enforce payment, without any reduction or allowance for the value of the shares at the time of forfeiture or for any consideration received on their disposal.
| 34. | Evidence of forfeiture |
A statutory declaration by a Director or the Secretary that a share has been forfeited on a specified date shall be conclusive evidence of the facts stated in it as against all persons claiming to be entitled to the share. The declaration shall (subject to the execution of an instrument of transfer if necessary) constitute a good title to the share. The person to whom the share is transferred or sold shall not be bound to see to the application of the purchase money or other consideration (if any), nor shall his or her title to the share be affected by any act, omission or irregularity relating to or connected with the proceedings in reference to the forfeiture or disposal of the share.
| 35. | Form of transfer |
| 35.1 | Subject to these Articles: |
| (a) | each member may transfer all or any of his or her shares which are in certificated form by instrument of transfer in writing in any usual form or in any form approved by the Board. Such instrument shall be executed by or on behalf of the transferor and (in the case of a transfer of a share which is not fully paid up) by or on behalf of the transferee. All instruments of transfer, when registered, may be retained by the Company; and |
| (b) | each member may transfer all or any of his or her shares which are in uncertificated form by means of a relevant system in such manner provided for, and subject as provided in, the uncertificated securities rules. No provision of these Articles shall apply in respect of an uncertificated share to the extent that it requires or contemplates the effecting of a transfer by an instrument in writing or the production of a certificate for the share to be transferred. |
| 35.2 | The transferor of a share shall be deemed to remain the holder of the share concerned until the name of the transferee is entered in the Register in respect of it. |
| 36. | Right to refuse registration of transfer |
| 36.1 | The Board may, in its absolute discretion, refuse to register any transfer of a share in certificated form (or renunciation of a renounceable letter of allotment) unless: |
| (a) | it is for a share which is fully paid up; |
| (b) | it is for a share upon which the Company has no lien; |
| (c) | it is only for one class of share; |
| (d) | it is in favour of a single transferee or no more than four joint transferees; |
| (e) | it is duly stamped or is duly certificated or otherwise shown to the satisfaction of the Board to be exempt from stamp duty (in each case if this is required); and |
| (f) | it is delivered for registration to the Office (or such other place as the Board may determine), accompanied (except in the case of a transfer by a person to whom the Company is not required by law to issue a certificate and to whom a certificate has not been issued or in the case of a renunciation) by the certificate for the shares to which it relates and such other evidence as the Board may reasonably require to prove the title of the transferor (or person renouncing) and the due execution of the transfer or renunciation by him or her or, if the transfer or renunciation is executed by some other person on his or her behalf, the authority of that person to do so. |
| 36.2 | The Board shall not refuse to register any transfer or renunciation of partly paid shares which are admitted to, or for which depositary instruments representing such shares are admitted to, Nasdaq on the grounds that they are partly paid shares in circumstances where such refusal would prevent dealings in such shares from taking place on an open and proper basis. |
| 36.3 | Transfers of shares will not be registered in the circumstances referred to in Article 74. |
| 36.4 | The Board may refuse to register a transfer of uncertificated shares in any circumstances that are allowed or required by the uncertificated securities rules and the relevant system. |
| 37. | Notice of refusal to register a transfer |
If the Board refuses to register a transfer of a share it shall notify the transferee of the refusal and the reasons for it within two months after the date on which the transfer was lodged with the Company or the instructions to the relevant system received. Any instrument of transfer which the Board refuses to register shall be returned to the person depositing it (except if there is suspected or actual fraud). All instruments of transfer which are registered may be retained by the Company.
| 38. | No fees on registration |
No fee shall be charged for registration of a transfer or other document or instruction relating to or affecting the title to any share or for making any other entry in the Register.
| 39. | Other powers in relation to transfers |
Nothing in these Articles shall prevent the Board:
| (a) | from recognising a renunciation of the allotment of any share by the allottee in favour of another person; or |
| (b) | (if empowered to do so by these Articles) from authorising any person to execute an instrument of transfer of a share and from authorising any person to transfer that share in accordance with any procedures implemented under Article 19. |
| 40. | Transmission of shares on death |
If a member dies, the survivors or survivor (where he or she was a joint holder), and his or her executors or administrators (where he or she was a sole or the only survivor of joint holders), shall be the only persons recognised by the Company as having any title to his or her shares. Nothing in these Articles shall release the estate of a deceased member from any liability for any share which has been solely or jointly held by him or her.
| 41. | Election of person entitled by transmission |
| 41.1 | Any person becoming entitled to a share because of the death or bankruptcy of a member, or otherwise by operation of law, may (on such evidence as to his or her title being produced as the Board may require) elect either to become registered as a member or to have some person nominated by him or her registered as a member. If he or she elects to become registered himself or herself, he or she shall notify the Company to that effect. If he or she elects to have some other person registered, he or she shall execute an instrument of transfer of such share to that person. All the provisions of these Articles relating to the transfer of shares shall apply to the notice or instrument of transfer (as the case may be) as if it were an instrument of transfer executed by the member and his or her death, bankruptcy or other event had not occurred. Where the entitlement of a person to a share because of the death or bankruptcy of a member or otherwise by operation of law is proved to the satisfaction of the Board, the Board shall within thirty days after proof cause the entitlement of that person to be noted in the Register. |
| 41.2 | A person entitled by transmission to a share in uncertificated form who elects to have some other person registered shall either: |
| (a) | procure that instructions are given by means of the relevant system to effect transfer of such uncertificated share to that person; or |
| (b) | change the uncertificated share to certificated form and execute an instrument of transfer of that certificated share to that person. |
| 42. | Rights on transmission |
Where a person becomes entitled to a share because of the death or bankruptcy of any member, or otherwise by operation of law, the rights of the holder in relation to such share shall cease. However, the person so entitled may give a good discharge for any dividends and other monies payable in respect of it and shall have the same rights to which he or she would be entitled if he or she were the holder of the share, except that he or she shall not be entitled to receive notice of, or to attend or vote at, any meeting of the Company or any separate meeting of the holders of any class of shares of the Company before he or she is registered as the holder of the share. The Board may at any time give notice requiring any such person to elect either to be registered himself or herself or to transfer the share. If the notice is not complied with within thirty days, the Board may withhold payment of all dividends and the other monies payable in respect of such share until the requirements of the notice have been complied with.
| 43. | Destruction of documents |
| 43.1 | The Company may destroy any: |
| (a) | instrument of transfer, after six years from the date on which it is registered; |
| (b) | dividend mandate or any variation or cancellation of a dividend mandate or any notification of change of name or address, after two years from the date on which it is recorded; |
| (c) | share certificate, after one year from the date on which it is cancelled; |
| (d) | instrument of proxy which has been used for the purpose of voting at any time after one year has elapsed from the date of use; |
| (e) | instrument of proxy which has not been used for the purpose of voting at any time after a period of one month has elapsed from the end of the meeting to which the instrument of proxy relates; |
| (f) | Share Warrant (including coupons or tokens detailed from it) which has been cancelled at any time after seven years from the date on which it was cancelled; or |
| (g) | other document for which any entry in the Register is made, after six years from the date on which an entry was first made in the Register in respect of it, |
provided that the Company may destroy any such type of document at a date earlier than that authorised by this Article if a copy of such document is made and retained (whether electronically, by microfilm, by digital imaging or by other similar means) until the expiration of the period applicable to the destruction of the original of such document.
| 43.2 | It shall be conclusively presumed in favour of the Company that every: |
| (a) | entry in the Register purporting to have been made on the basis of a document so destroyed was duly and properly made; |
| (b) | instrument of transfer so destroyed was duly registered; |
| (c) | share certificate so destroyed was duly cancelled; and |
| (d) | other document so destroyed had been properly dealt with under its terms and was valid and effective according to the particulars in the records of the Company. |
| 43.3 | This Article shall only apply to the destruction of a document in good faith and without notice of any claim (regardless of the parties to it) to which the document might be relevant. Nothing in this Article shall be construed as imposing any liability on the Company in respect of the destruction of any such document other than as provided for in this Article which would not attach to the Company in the absence of this Article. References in this Article to the destruction of any document include references to the disposal of it in any manner. |
| 43.4 | References in this Article to instruments of transfer shall include, in relation to uncertificated shares, instructions and/or notifications made in accordance with the relevant system relating to the transfer of such shares. |
| 44. | Sub-division |
Any resolution authorising the Company to sub-divide its shares or any of them may determine that, as between the shares resulting from the sub-division, any of them may have any preference or advantage or be subject to any restriction as compared with the others.
| 45. | Fractions |
| 45.1 | Where any difficulty arises in regard to any consolidation or division, the Board may settle such difficulty as they see fit. In particular, without limitation, the Directors may sell to any person (including the Company) the shares representing the fractions for the best price reasonably obtainable and distribute the net proceeds of sale in due proportion among those members in proportion to their fractional entitlements or retain such net proceeds for the benefit of the Company and: |
| (a) | in the case of shares in certificated form, the Board may authorise any person to execute an instrument of transfer of the shares to the purchaser or a person nominated by the purchaser and take such other steps (including the giving of directions to or on behalf of the holder, who shall be bound by them) as they think fit to effect such transfer; and |
| (b) | in the case of shares in uncertificated form, the Board may: |
| (i) | to enable the Company to deal with the share in accordance with the provisions of this Article, require or procure any relevant person or the Operator (as applicable) to convert the share into certificated form; and |
| (ii) | after such conversion, authorise any person to execute an instrument of transfer of the shares to the purchaser or a person nominated by the purchaser and take such other steps (including the giving of directions to or on behalf of the holder, who shall be bound by them) as they think fit to effect the transfer. |
| 45.2 | The transferee shall not be bound to see to the application of the purchase money nor shall his or her title to the shares be affected by any irregularity in or invalidity of the proceedings in reference to the sale. |
| 46. | Annual general meetings |
An annual general meeting shall be held once a year, at such time (consistent with the terms of the Companies Acts) and place, including partly or wholly by means of electronic facility or facilities, as may be determined by the Board.
| 47. | Convening and format of general meetings |
| 47.1 | All meetings other than annual general meetings shall be called general meetings. The Board or the chair of the Board may, whenever it, he or she thinks fit, and shall on requisition in accordance with the Companies Acts, proceed to convene a general meeting. For all other purposes, and unless expressly provided otherwise in these Articles, the procedures for giving notice (other than as to duration) of, the conduct of, and voting at annual general meetings and all other general meetings shall be the same. In the case of a general meeting called pursuant to a requisition under the Companies Acts, unless such meeting shall have been called by the Directors, no business other than that stated in the requisition as the object of the meeting shall be discussed. |
| 47.2 | The Directors may make whatever arrangements they consider fit to allow those entitled to do so to attend and participate in any general meeting. The Directors shall determine in relation to each general meeting the means of attendance at and participation in the general meeting, including whether the persons entitled to attend and participate in the general meeting shall be enabled to do so: |
| (a) | by simultaneous attendance and participation at a satellite place or places pursuant to Article 47.3; and/or |
| (b) | by means of electronic facility or facilities pursuant to Article 47.4 or Article 47.5, |
(and for the avoidance of doubt, the Directors shall be under no obligation to offer or provide such satellite place or places or such electronic facility or facilities).
| 47.3 | In the case of any general meeting and without prejudice to Article 47.4 and Article 47.5, the Directors may make arrangements for simultaneous attendance at and participation in the general meeting in more than one physical place anywhere in the world by persons entitled to attend the meeting. The members present in person or by proxy at a satellite place shall be counted in the quorum for, and be entitled to vote at, the general meeting in question. The general meeting shall be duly constituted and its proceedings valid if the chair of the general meeting is satisfied that adequate facilities are available throughout the meeting to ensure that members attending at the principal place and any satellite place(s) (each as defined below) are able to: |
| (a) | participate in the business for which the meeting has been convened; |
| (b) | hear all persons who speak (whether by the use of microphones, loudspeakers, audio-visual communications equipment or otherwise) in the principal meeting place and any satellite meeting place; and |
| (c) | be heard by all other persons so present in the same way. |
The general meeting shall be deemed to take place at the place where the chair of the general meeting presides (the “principal place”, with any other location where that meeting takes place being referred in these Articles as a “satellite place”). The powers of the chair shall apply equally to each satellite place, including his or her power to adjourn the meeting as referred to in Article 56.
| 47.4 | Without prejudice to Article 47.3 and Article 47.5, the Directors may determine in relation to any general meeting (including any general meeting that is being held at more than one physical place) to enable persons entitled to attend and participate to do so by simultaneous attendance and participation by means of electronic facility or facilities determined by the Directors (any such general meeting being a “hybrid general meeting”). The members or their proxies present personally or by means of an electronic facility or facilities shall be counted in the quorum for, and entitled to participate in, the general meeting in question. The general meeting shall be duly constituted and its proceedings valid if the chair of the general meeting is satisfied that adequate facilities are available throughout the general meeting to ensure that members attending the general meeting by all means (including by means of an electronic facility or facilities) are able to: |
| (a) | participate in the business for which the general meeting has been convened; |
| (b) | hear all persons who speak at the general meeting; and |
| (c) | be heard by all other persons attending and participating in the general meeting. |
| 47.5 | Without prejudice to Article 47.3 and Article 47.4, the Directors may determine in relation to any general meeting to enable persons entitled to attend and participate to do so by means of electronic facility or facilities determined by the Directors with no member necessarily in physical attendance (any such general meeting being an “electronic general meeting”). The members or their proxies present by means of an electronic facility or facilities shall be counted in the quorum for, and entitled to participate in, the general meeting in question. The general meeting shall be duly constituted and its proceedings valid if the chair of the general meeting is satisfied that adequate facilities are available throughout the general meeting to ensure that members attending the general meeting who are not present together at the same place may, by means of an electronic facility or facilities, attend, speak and vote at it. |
| 47.6 | If a general meeting is held as a hybrid general meeting or an electronic general meeting, the Directors (and, at a general meeting, the chair) may (subject to the requirements of Companies Acts) make any arrangement and impose any requirement or restriction in connection with participation by such electronic facility or facilities, including any arrangement, requirement or restriction that is: |
| (a) | necessary to ensure the identification of those taking part and the security of the electronic facility or facilities; and |
| (b) | proportionate to the achievement of those objectives. |
In this respect, the Board may authorise any voting application, system or facility for hybrid general meetings or electronic general meetings as it sees fit.
| 47.7 | If, at any hybrid general meeting or electronic general meeting, any document is required to be on display or to be available for inspection at the meeting (whether prior to or for the duration of the meeting or both), the Company shall ensure that it is available in electronic form to persons entitled to inspect it for at least the required period of time, and this will be deemed to satisfy any such requirement. |
| 47.8 | Nothing in these Articles: |
| (a) | shall preclude the holding and conducting of a general meeting in such a way that persons who are not present together at the same place may by electronic means attend and speak and vote at it; or |
| (b) | prevents a general meeting being held both physically and electronically. |
| 48. | Notice of general meetings |
A general meeting shall be called by at least such minimum notice as is required or permitted by the Companies Acts. The period of notice shall in either case be exclusive of the day on which it is served or deemed to be served and of the day on which the meeting is to be held and shall be given to all members other than those who are not entitled to receive such notices from the Company. The Company may give such notice by any means or combination of means permitted by the Companies Acts.
| 49. | Contents of notice of general meetings |
| 49.1 | Every notice calling a general meeting shall specify; |
| (a) | whether the meeting shall be a physical general meeting, a hybrid general meeting or an electronic general meeting; |
| (b) | in the case of a physical general meeting, the time, date and place (including any satellite place or places determined pursuant to Article 47.3, which shall be identified as such in the notice) of the meeting; |
| (c) | in the case of a hybrid general meeting, the time, date and place of the meeting (including any satellite place or places determined pursuant to Article 47.3, which shall be identified as such in the notice) and the electronic facility or facilities for the meeting, which electronic facility or facilities may vary from time to time and from meeting to meeting as the Board in its sole discretion sees fit; and |
| (d) | in the case of an electronic general meeting, the time, date and electronic facility or facilities for the meeting, which electronic facility or facilities may vary from time to time and from meeting to meeting as the Board in its sole discretion sees fit. |
There shall appear with reasonable prominence in every such notice a statement that a member entitled to attend and vote is entitled to a proxy or (if he, she or it has more than one share) proxies to exercise all or any of his, her or its rights to attend, speak and vote and that a proxy need not be a member of the Company. Such notice shall also include the address of the website on which the information required by the Act is published, state the procedures with which members must comply in order to be able to attend and vote at the meeting (including the date by which they must comply), provide details of any forms to be used for the appointment of a proxy and state that a member has the right to ask questions at the meeting in accordance with the Act.
| 49.2 | The notice shall specify the general nature of the business to be transacted at the meeting and shall set out the text of all resolutions to be considered by the meeting and shall state in each case whether it is proposed as an ordinary resolution or as a special resolution. |
| 49.3 | In the case of an annual general meeting, the notice shall also specify the meeting as such. |
| 49.4 | For the purposes of determining which persons are entitled to attend or vote at a meeting and how many votes a person may cast, the Company may specify in the notice of meeting a time, not more than forty-eight hours before the time fixed for the meeting (not taking into account non-working days) by which a person must be entered in the Register in order to have the right to attend or vote at the meeting or appoint a proxy to do so. |
| 50. | Omission to give notice and non-receipt of notice |
The accidental omission to give notice of any meeting or to send an instrument of proxy (where this is intended to be sent out with the notice) to, or the non-receipt of either by, any person entitled to receive the same shall not invalidate the proceedings of that meeting.
| 51. | Postponement of general meeting |
If, after the sending of the notice of a general meeting but before the meeting is held, or after the adjournment of a general meeting but before the adjourned meeting is held (whether or not notice of the adjourned meeting is required), the Board, in its absolute discretion, considers that it is impracticable or unreasonable for any reason to hold a physical general meeting or hybrid general meeting at the declared place (or any of the declared places, in the case of a satellite meeting) and/or a hybrid general meeting or electronic general meeting by means of the electronic facility or facilities specified in the notice on the date or at the time stated in the notice calling the meeting, it may change the place (or any of the places, in the case of a satellite meeting) and/or electronic facility or facilities and/or postpone the time and/or date at which the meeting is to be held (or do any of the foregoing). The Board shall take reasonable steps to ensure that notice of the date, time and place of, and/or the electronic facility or facilities for, the rearranged meeting is given to any member trying to attend the meeting at the original time and at the original place (or places, in the case of a satellite meeting) and/or through the original electronic facility or facilities. Where a general meeting is so postponed, notice of the date, time and place of, and/or the electronic facility or facilities for, the rearranged meeting shall, if practicable, also be placed in at least two national newspapers published in the United Kingdom. Notice of the business to be transacted at such rearranged meeting shall not be required, provided that it is the same as the business which might properly have been transacted at the meeting had it not been rearranged. If a meeting is rearranged in accordance with this Article 51, appointments of proxy will be valid if they are received as required by these Articles not less than forty-eight hours before the time appointed for holding the rearranged meeting and for the purpose of calculating this period, the Board can decide in their absolute discretion, not to take account of any part of a day that is not a working day. The Board may also postpone or move the rearranged meeting (or do both) under this Article.
| 52. | Quorum at general meeting |
No business shall be transacted at any general meeting unless a quorum is present. If a quorum is not present a chair of the meeting can still be chosen and this will not be treated as part of the business of the meeting. Two members present in person or by proxy and entitled to attend and to vote on the business to be transacted shall be a quorum.
| 53. | Procedure if quorum not present |
If a quorum is not present within fifteen minutes (or such longer interval as the chair in his or her absolute discretion thinks fit) from the time appointed for holding a general meeting, or if a quorum ceases to be present during a meeting, the meeting shall be dissolved if convened on the requisition of members. In any other case, the meeting shall stand adjourned to another day (not being less than ten clear days after the date of the original meeting), and at such time and place or places and/or by means of such electronic facility or facilities, as the chair (or, in default, the Board) may determine. If at such adjourned meeting a quorum is not present within fifteen minutes from the time appointed for holding the meeting, one person entitled to vote on the business to be transacted, being a member or a proxy for a member or a duly authorised representative of a corporation which is a member, shall be a quorum and any notice of an adjourned meeting shall state this.
| 54. | Chair of general meeting |
The chair of the Board shall preside at every general meeting of the Company. If there is no such chair or if at any meeting he or she shall not be present within five minutes after the time appointed for holding the meeting, or shall be unwilling to act as chair, the deputy chair (if any) of the Board shall, if present and willing to act, preside at such meeting. If more than one deputy chair is present they shall agree amongst themselves who is to take the chair or, if they cannot agree, the deputy chair who has been in office as a Director the longest shall take the chair. If no chair or deputy chair shall be so present and willing to act, the Directors present shall choose one of their number to act or, if there be only one Director present, he or she shall be chair if willing to act. If there be no Director present and willing to act, the members present and entitled to vote shall choose one of their number to be chair of the meeting. Nothing in these Articles shall restrict or exclude any of the powers or rights of a chair of a meeting which are given by law.
| 55. | Entitlement to attend, speak and participate |
| 55.1 | A Director (and any other person invited by the chair to do so) may attend and speak at any general meeting and at any separate meeting of the holders of any class of shares of the Company, whether or not he or she is also a member. |
| 55.2 | In relation to a physical general meeting, the right of a member who is entitled to attend and participate to participate in the business of any general meeting shall include, without limitation, the right to speak, vote on a poll, be represented by a proxy and have access to all documents which are required by the Companies Acts or these Articles to be made available at the meeting. |
| 55.3 | In relation to a hybrid general meeting or an electronic general meeting, the right of a member who is entitled to attend and participate to participate in the business of any general meeting shall include, without limitation, the right to speak, vote on a poll, be represented by a proxy and have access (including electronic access) to all documents which are required by the Companies Acts or these Articles to be made available at the meeting. |
| 55.4 | All persons seeking to attend and participate in a hybrid general meeting or an electronic general meeting by way of electronic facility or facilities shall be responsible for maintaining adequate facilities to enable them to do so. In no circumstances shall the inability of one or more members to access, or to continue to access, the electronic facility or facilities for participation in the meeting for all or part of the meeting affect the validity of the meeting or any business conducted at a hybrid general meeting or an electronic general meeting, provided that sufficient members are able to participate in the meeting as are required to constitute a quorum under Article 52. |
| 56. | Adjournments |
| 56.1 | The chair may, with the consent of a meeting at which a quorum is present, and shall, if so directed by the meeting, adjourn any meeting from time to time (or indefinitely) and from place to place (or, in the case of a meeting held at a principal place and one or more satellite places, such other places) and/or from such electronic facility or facilities for attendance and participation to such other electronic facility or facilities as determined by the chair in his or her absolute discretion. |
| 56.2 | Without prejudice to any other power which the chair may have under these Articles or at common law, he or she may, without the need for the consent of the meeting and before or after it has started, interrupt or adjourn any meeting from time to time and from place to place (or places in the case of a meeting to which Article 47.3 applies) or from electronic facility or facilities to electronic facility or facilities, or for an indefinite period, if he or she is of the opinion that it has become necessary to do so in order: |
| (a) | to secure the proper and orderly conduct of the meeting; or |
| (b) | to give all persons entitled to do so a reasonable opportunity of attending, speaking and voting at the meeting; or |
| (c) | to ensure that the business of the meeting is properly disposed of. |
| 56.3 | If it appears to the chair that the facilities at the principal place or any satellite place or an electronic facility or facilities or security at any general meeting have become inadequate for the purposes referred to in Articles 47.3, 47.4 or 47.5 (as applicable) or are otherwise not sufficient to allow the meeting to be conducted substantially in accordance with the provisions set out in the notice of meeting, then the chair may, without the consent of the meeting, interrupt or adjourn the general meeting. |
| 56.4 | All business conducted at a meeting up to the time of any adjournment shall, subject to Article 56.5, be valid. |
| 56.5 | The chair may specify that only the business conducted at the meeting up to a point in time which is earlier than the time of the adjournment is valid, if in his or her opinion, to do so would be more appropriate. |
| 56.6 | Meetings can be adjourned more than once, in accordance with the procedures set out in this Article 56. |
| 57. | Notice of adjournment |
If the meeting is adjourned indefinitely or for more than three months, notice of the adjourned meeting shall be given in the same manner as in the case of the original meeting. Subject to the provisions of the Companies Acts and the provisions of Article 53, if notice of an adjourned meeting is required in accordance with this Article 57, such notice shall be sent at least seven (7) clear days before the date of the adjourned meeting specifying the date, time and place or electronic facility of the adjourned meeting and the general nature of the business to be transacted. Except as provided in these Articles, there is no need to give notice of the adjourned meeting or of the business to be considered there.
| 58. | Business of adjourned meeting |
No business shall be transacted at any adjourned meeting other than the business which might properly have been transacted at the meeting from which the adjournment took place.
| 59. | Security arrangements and orderly conduct |
| 59.1 | The Board may, for the purpose of controlling the level of attendance or ensuring the safety of those attending at any place specified for the holding of a physical general meeting or hybrid general meeting, ensuring the security of the meeting and ensuring the future orderly conduct of the meeting, from time to time make such arrangements as it shall in its absolute discretion consider to be appropriate and may from time to time vary any such arrangements or make new arrangements therefor. Any decision made under this Article 59.1 shall be final and the entitlement of any member or proxy to attend a general meeting at such place (or places, in the case of a meeting to which Article 47.3 applies) shall be subject to any such arrangements as may be for the time being approved by the Board. |
| 59.2 | The Board may direct that any person wishing to attend any general meeting should provide such evidence of identity and submit to such searches or other security arrangements or restrictions as the Board shall consider appropriate in the circumstances and shall be entitled in its absolute discretion to refuse entry to any general meeting to any person who fails to provide such evidence of identity or to submit to such searches or to otherwise comply with such security arrangements or restrictions. |
| 59.3 | The Board shall be entitled in its absolute discretion to authorise one or more persons (including the Directors, the Secretary or the chair) to refuse physical or electronic entry to, or eject (physically or electronically) from, any meeting any person who fails to provide such evidence of identity or to submit to such searches or to otherwise comply with such security arrangements or restrictions as are required pursuant to this Article, or who causes the meeting to become disorderly. |
| 59.4 | Subject to the Act (and without prejudice to any other powers vested in the chair of a meeting), the chair shall take such action or give such directions as he or she thinks fit to promote the orderly conduct of the business of the meeting as laid down in the notice of the meeting and in the case of a physical general meeting or a hybrid general meeting to ensure the security of the meeting and the safety of the people attending the meeting. The chair’s decision on points of order, matters of procedure on or matters arising incidentally from the business of the meeting shall be final, as shall be his or her determination as to whether any matter is of such a nature. |
| 60. | Overflow meeting rooms |
| 60.1 | The Board may, in accordance with this Article, make arrangements for members and proxies who are entitled to attend and participate in a physical general meeting or a hybrid general meeting, but who cannot be seated in the main meeting room where the chair will be, to attend and take part in a general meeting in an overflow room or rooms. Any overflow room will have appropriate links to the main room and will enable audio-visual communication between the meeting rooms throughout the meeting. The Board will decide how to divide members and proxies between the main room and the overflow room. If an overflow room is used, the meeting will be treated as being held and taking place in the main meeting room and the meeting will consist of all the members and proxies who are attending both in the main meeting room and the overflow room. |
| 60.2 | Details of any arrangements for overflow rooms will be set out in the notice of the meeting but failure to do so will not invalidate the meeting. |
| 60.3 | The Board may make arrangements for members and proxies who are entitled to attend and participate in a physical general meeting or a hybrid general meeting or an adjourned general meeting, to be able to view and hear the proceedings of the general meeting or adjourned general meeting and to speak at the meeting (whether by use of microphones, loudspeakers, audio-visual communications equipment or otherwise) by attending at a venue anywhere in the world not being a satellite meeting place. If the general meeting is only held as a physical general meeting and not also a hybrid general meeting, those attending at any such venue shall not be regarded as present at the general meeting or adjourned general meeting and shall not be entitled to vote as the meeting at or from that venue. The inability for any reason of any member present in person or by proxy at such a venue to view or hear all or any of the proceedings of the physical general meeting or to speak at the meeting shall not in any way affect the validity of the proceedings of the meeting. |
| 61. | Amendment to resolutions |
| 61.1 | If an amendment to any resolution under consideration is proposed but is ruled out of order by the chair of the meeting in good faith, any error in such ruling shall not invalidate the proceedings on the original resolution. |
| 61.2 | In the case of a resolution duly proposed as a special resolution, no amendment to it (other than an amendment to correct a patent error) may in any event be considered or voted on. In the case of a resolution duly proposed as an ordinary resolution no amendment to it (other than an amendment to correct a patent error) may be considered or voted on unless either at least forty-eight hours prior to the time appointed for holding the meeting or adjourned meeting at which such ordinary resolution is to be proposed, notice in writing of the terms of the amendment and intention to move the same has been lodged at the Office or received in electronic form at the electronic address at which the Company has or is deemed to have agreed to receive it or the chair of the meeting in his or her absolute discretion decides that it may be considered or voted on. |
| 62. | Withdrawal and ruling amendments out of order |
With the consent of the chair of the meeting, an amendment may be withdrawn by its proposer before it is voted on. If an amendment proposed to any resolution under consideration is ruled out of order by the chair of the meeting, the proceedings on the resolution shall not be invalidated by any error in the ruling.
| 63. | Members’ resolutions |
| 63.1 | Members of the Company shall have the rights provided by the Companies Acts to have the Company circulate and give notice of a resolution which may be properly moved, and is intended to be moved, at the Company’s next annual general meeting. |
| 63.2 | Expenses of complying with these rights shall be borne in accordance with the Companies Acts. |
| 64. | Method of voting |
| 64.1 | Any resolution put to the vote of a general meeting must be decided exclusively on a poll. |
| 64.2 | At general meetings, resolutions shall be put to the vote by the chair of the meeting and there shall be no requirement for the resolution to be proposed or seconded by any person. |
| 65. | Objection to error in voting |
No objection shall be raised to the qualification of any voter or to the counting of, or failure to count, any vote, except at the meeting or adjourned meeting at which the vote objected to is given or tendered or at which the error occurs. Any objection or error shall be referred to the chair of the meeting and shall only vitiate the decision of the meeting on any resolution if the chair decides that the same is of sufficient magnitude to vitiate the resolution or may otherwise have affected the decision of the meeting. The decision of the chair of the meeting on such matters shall be final and conclusive.
| 66. | Voting procedure |
| 66.1 | Any poll on any question of adjournment shall be taken immediately. A poll on any other matter shall be taken in such manner (including the use of ballot or voting papers or tickets or electronic means, or any combination thereof) and at such time and place, not more than thirty days from the date of the meeting or adjourned meeting, as the chair shall direct. The chair may appoint scrutineers who need not be members. It is not necessary to give notice of a poll not taken immediately if the time and place at which it is to be taken are announced at the meeting. In any other case, at least seven clear days’ notice shall be given specifying the time, date and place at which the poll shall be taken. The result of the poll shall be deemed to be the resolution of the meeting at which the poll was due to be conducted. |
| 66.2 | Votes may be given in person or by proxy. A member entitled to more than one vote need not, if he, she or it votes, use all his, her or its votes or cast all the votes he, she or it uses in the same way. |
| 66.3 | No notice need be given of a poll not taken during the meeting if the time and place at which it is to be taken are announced at the meeting. In any other case, at least seven clear days’ notice must be given specifying the time and place at which the poll is to be taken. |
| 67. | Votes of members |
| 67.1 | Subject to Article 67.2, to the Companies Acts and to any special terms as to voting on which any shares may have been issued or may for the time being be held (including, without limitation, in respect of the Deferred Shares and the Non-Voting Ordinary Shares, Article 6) and to any suspension or abrogation of voting rights under these Articles, at any general meeting: |
| (a) | every member who is present in person or by duly appointed proxy or corporate representative has one vote for every share of which he, she or it is the holder or in respect of which his, her or its appointment as proxy or corporate representative has been made; and |
| (b) | a member, proxy or corporate representative entitled to more than one vote need not, if he, she or it votes, use all his, her or its votes or cast all the votes he, she or it uses the same way. |
| 67.2 | If two or more persons are joint holders of a share, then in voting on any question the vote of the senior who tenders a vote, whether in person or by proxy, shall be accepted to the exclusion of the votes of the other joint holders. For this purpose seniority shall be determined by the order in which the names of the holders stand in the Register. |
| 67.3 | Where in England or elsewhere a receiver or other person (by whatever name called) has been appointed by any court claiming jurisdiction in that behalf to exercise powers with respect to the property or affairs of any member on the ground (however formulated) of mental disorder, the Board may in its absolute discretion, upon or subject to production of such evidence of the appointment as the Board may require, permit such receiver or other person on behalf of such member to vote in person by proxy on behalf of such member at any general meeting or to exercise any other right conferred by membership in relation to meetings of the Company. Evidence to the satisfaction of the Board of the authority of the person claiming to exercise the right to vote shall be deposited at the Office, or at such other place as is specified in accordance with these Articles for the deposit of instruments of proxy, at least forty-eight hours before the time appointed for holding the meeting or adjourned meeting at which the right to vote is to be exercised and, in default, the right to vote shall not be exercisable. |
| 67.4 | In the case of equality of votes the chair of the meeting shall not be entitled to a casting vote. |
| 68. | No right to vote where sums overdue on shares |
No member may vote at a general meeting (or any separate meeting of the holders of any class of shares), either in person or by proxy, or to exercise any other right or privilege as a member in respect of a share held by him or her unless:
| (a) | all calls or other sums presently due and payable by him or her in respect of that share whether alone or jointly with any other person together with interest and expenses (if any) have been paid to the Company; or |
| (b) | the Board determines otherwise. |
| 69. | Voting by Proxy |
| 69.1 | Subject to Article 69.2, an instrument appointing a proxy shall be in writing in any usual form (or in another form approved by the Board) executed under the hand of the appointor or his or her duly constituted attorney or, if the appointor is a corporation, under its seal or signed by a duly authorised officer or attorney or other person authorised to sign. |
| 69.2 | Subject to the Companies Acts, the Board may accept the appointment of a proxy received by electronic means on such terms and subject to such conditions as it considers fit. The appointment of a proxy received by electronic means shall not be subject to the requirements of Article 69.1. |
| 69.3 | For the purposes of Articles 69.1 and 69.2, the Board may require such reasonable evidence it considers necessary to determine: |
| (a) | the identity of the member and the proxy; and |
| (b) | where the proxy is appointed by a person acting on behalf of the member, the authority of that person to make the appointment. |
| 69.4 | A member may appoint another person as his or her proxy to exercise all or any of his or her rights to attend and to speak and to vote on a resolution or amendment of a resolution, or on other business arising, at a meeting or meetings of the Company. Unless the contrary is stated in it, the appointment of a proxy shall be deemed to confer authority to exercise all such rights, as the proxy thinks fit. |
| 69.5 | A proxy need not be a member. |
| 69.6 | A member may appoint more than one proxy in relation to a meeting, provided that each proxy is appointed to exercise the rights attached to different shares held by the member. When two or more valid but differing appointments of proxy are delivered or received for the same share for use at the same meeting, the one which is last validly delivered or received (regardless of its date or the date of its execution) shall be treated as replacing and revoking the other or others as regards that share. If the Company is unable to determine which appointment was last validly delivered or received, none of them shall be treated as valid in respect of that share. |
| 69.7 | Delivery or receipt of an appointment of proxy does not prevent a member attending and voting in person at the meeting or an adjournment of the meeting. |
| 69.8 | The appointment of a proxy shall (unless the contrary is stated in it) be valid for an adjournment of the meeting as well as for the meeting or meetings to which it relates. The appointment of a proxy shall be valid for twelve months from the date of execution or, in the case of an appointment of proxy delivered by electronic means, for twelve months from the date of delivery unless otherwise specified by the Board. |
| 69.9 | Subject to the Companies Acts, the Company may send a form of appointment of proxy to all or none of the persons entitled to receive notice of and to vote at a meeting. If sent, the form shall provide for three-way voting on all resolutions (other than procedural resolutions) set out in the notice of meeting. |
| 69.10 | The Company shall not be bound to enquire whether any proxy or corporate representative votes in accordance with the instructions given to him, her or it by the member he, she or it represents and if a proxy or corporate representative does not vote in accordance with the instructions of the member he, she or it represents the vote or votes cast shall nevertheless be valid for all purposes. |
| 70. | Receipt of proxy |
| 70.1 | An instrument appointing a proxy and any reasonable evidence required by the Board in accordance with Article 69.3 shall: |
| (a) | subject to Articles 70.1(c) and (d), in the case of an instrument of proxy in hard copy form, delivered to the Office, or another place in the United Kingdom specified in the notice convening the meeting or in the form of appointment of proxy or other accompanying document sent by the Company in relation to the meeting (a “proxy notification address”) not less than forty-eight hours before the time for holding the meeting or adjourned meeting at which the person named in the form of appointment of proxy proposes to vote or by such later time as is specified in the notice or instrument; |
| (b) | subject to Articles 70.1(c) and (d), in the case of an appointment of a proxy sent by electronic means, where the Company has given an electronic address (a “proxy notification electronic address”): |
| (i) | in the notice calling the meeting; |
| (ii) | in an instrument of proxy sent out by the Company in relation to the meeting; |
| (iii) | in an invitation to appoint a proxy issued by the Company in relation to the meeting; or |
| (iv) | on a website maintained by or on behalf of the Company on which any information relating to the meeting is required by the Act to be kept, |
it shall be received at such proxy notification electronic address not less than forty-eight hours before the time for holding the meeting or adjourned meeting at which the person named in the form of appointment of proxy proposes to vote or by such later time as is specified in any of the methods of notice in (i) to (iv) above.
| (c) | in the case of a poll taken more than forty-eight hours after it is demanded, delivered or received at a proxy notification address or a proxy notification electronic address and not less than twenty-four hours before the time appointed for the holding of the adjourned meeting; or |
| (d) | in the case of a poll which is not taken at the meeting but is taken forty-eight hours or less thereafter, or in the case of an adjourned meeting to be held forty-eight hours or less after the time fixed for holding the original meeting, received: |
| (i) | at a proxy notification address or a proxy notification electronic address in accordance with Articles 70.1(a) or (b); |
| (ii) | by the chair of the meeting or the secretary or any Director at the meeting, as the case may be, at the original meeting; or |
| (iii) | at a proxy notification address or a proxy notification electronic address by such time as the chair of the meeting may direct at the meeting. |
In calculating the periods in this Article, no account shall be taken of any part of a day that is not a working day.
| 70.2 | The Board may decide, either generally or in any particular case, to treat a proxy appointment as valid notwithstanding that the appointment or any of the information required under Article 69.3 has not been received in accordance with the requirements of this Article. |
| 70.3 | Subject to Article 70.2, if the proxy appointment and any of the information required under Article 69.3 is not received in the manner set out in Article 70.1, the appointee shall not be entitled to vote in respect of the shares in question. |
| 70.4 | Without limiting the foregoing, in relation to any uncertificated shares, the Board may from time to time: |
| (a) | permit appointments of a proxy by means of a communication sent in electronic form in the form of an uncertificated proxy instruction; and |
| (b) | permit supplements to, or amendments or revocations of, any such uncertificated proxy instruction by the same means. |
The Board may in addition prescribe the method of determining the time at which any such uncertificated proxy instruction is to be treated as received by the Company or a participant acting on its behalf. The Board may treat any such uncertificated proxy instruction which purports to be or is expressed to be sent on behalf of a holder of a share as sufficient evidence of the authority of the person sending that instruction to send it on behalf of that holder.
| 71. | Revocation of proxy |
A vote given shall be valid in the event of the death or mental disorder of the principal or the revocation of the instrument of proxy, or of the authority under which the instrument of proxy was executed, or the transfer of the share for which the instrument of proxy is given, unless notice in writing of such death, mental disorder, revocation or transfer shall have been received by the Company at the Office, or at such other place as has been appointed for the deposit of instruments of proxy, no later than the last time at which an appointment of a proxy should have been received in order for it to be valid for use at the meeting at which the vote was given.
| 72. | Availability of appointments of proxy |
| 72.1 | The Directors may at the expense of the Company send or make available appointments of proxy or invitations to appoint a proxy to the members by post or by electronic means or otherwise (with or without provision for their return prepaid) for use at any general meeting or at any separate class meeting, either in blank or nominating in the alternative any one or more of the Directors or any other person. |
| 72.2 | If for the purpose of any meeting, appointments of proxy or invitations to appoint as proxy a person or one of a number of persons specified in the invitations are issued at the Company’s expense, they shall be issued to all (and not to some only) of the members entitled to be sent a notice of the meeting and to vote at it. The accidental omission, or the failure due to circumstances beyond the Company’s control, to send or make available such an appointment of proxy or give such an invitation to, or the non-receipt thereof by, any member entitled to attend and vote at a meeting shall not invalidate the proceedings at that meeting. |
| 73. | Corporate representatives |
| 73.1 | A corporation (whether or not a company within the meaning of the Act) which is a member may, by resolution of its Directors or other governing body, authorise such person as it thinks fit to act as its representative (or, as the case may be, representatives) at any meeting of the Company or at any separate meeting of the holders of any class of shares. |
| 73.2 | Any person so authorised shall be entitled to exercise the same powers on behalf of the corporation (in respect of that part of the corporation’s holdings to which the authority relates) as the corporation could exercise if it were an individual member. |
| 73.3 | The corporation shall for the purposes of these Articles be deemed to be present in person and at any such meeting if a person so authorised is present at it, and all references to attendance and voting in person shall be construed accordingly. |
| 73.4 | A Director, the Secretary or some person authorised for the purpose by the Secretary may require the representative to produce a certified copy of the resolution so authorising him or her or such other evidence of his or her authority reasonably satisfactory to them before permitting him or her to exercise his or her powers. |
| 73.5 | A vote given by a corporate representative shall be valid notwithstanding that he, she or it is no longer authorised to represent the member unless notice of the revocation of appointment was delivered in writing to the Company at such place or address and by such time as is specified in Article 71 for the revocation of the appointment of a proxy. |
| 74. | Failure to disclose interests in shares |
| 74.1 | If a member, or any other person appearing to be interested in shares held by that member, has been issued with a notice under section 793 of the Act (section 793 notice) and has failed in relation to any shares (default shares, which expression includes any shares issued after the date of such notice in right of those shares) to give the Company the information required by the section 793 notice within the prescribed period from the service of the notice, the following sanctions shall apply unless the Board determines otherwise: |
| (a) | the member shall not be entitled in respect of the default shares to be present or to vote (either in person or by representative or proxy) at any general meeting or at any separate meeting of the holders of any class of shares or to exercise any other right conferred by membership in relation to any such meeting; and |
| (b) | where the default shares represent at least 0.25% in nominal value of the issued shares of their class (calculated exclusive of any shares held as treasury shares): |
| (i) | any dividend or other money payable for such shares shall be withheld by the Company, which shall not have any obligation to pay interest on it, and the member shall not be entitled to elect, pursuant to Article 133, to receive shares instead of that dividend; and |
| (ii) | no transfer, other than an excepted transfer, of any shares held by the member shall be registered unless the member himself or herself is not in default of supplying the required information and the member proves to the satisfaction of the Board that no person in default of supplying such information is interested in any of the shares that are the subject of the transfer. |
| (c) | For the purposes of ensuring Article 74.1(b)(ii) can apply to all shares held by the member, the Company may in accordance with the uncertificated securities rules, issue a written notification to the Operator requiring conversion into certificated form of any share held by the member in uncertificated form. |
| 74.2 | Where the sanctions under Article 74.1 apply in relation to any shares, they shall cease to have effect (and any dividends withheld under Article 74.1(b) shall become payable): |
| (a) | if the shares are transferred by means of an excepted transfer but only in respect of the shares transferred; or |
| (b) | at the end of the period of seven days (or such shorter period as the Board may determine) following receipt by the Company of the information required by the section 793 notice and the Board being fully satisfied that such information is full and complete. |
| 74.3 | Where, on the basis of information obtained from a member in respect of any share held by him or her, the Company issues a section 793 notice to any other person, it shall at the same time send a copy of the notice to the member, but the accidental omission to do so, or the non-receipt by the member of the copy, shall not invalidate or otherwise affect the application of Article 74.1. |
| 74.4 | For the purposes of this Article: |
| (a) | a person, other than the member holding a share, shall be treated as appearing to be interested in that share if the member has informed the Company that the person is, or may be, so interested, or if the Company (after taking account of any information obtained from the member or, pursuant to a section 793 notice, from anyone else) knows or has reasonable cause to believe that the person is, or may be, so interested; |
| (b) | interested shall be construed as it is for the purpose of section 793 of the Act; |
| (c) | reference to a person having failed to give the Company the information required by a notice, or being in default as regards supplying such information, includes reference: |
| (i) | to his, her or it having failed or refused to give all of any part of it; and |
| (ii) | to his, her or it having given information which he or she knows to be false in a material particular or having recklessly given information which is false in a material particular; |
| (d) | prescribed period means fourteen days; |
| (e) | excepted transfer means, in relation to any shares held by a member: |
| (i) | a transfer by way of or pursuant to acceptance of a takeover offer for the Company (within the meaning of section 974 of the Act); or |
| (ii) | a transfer in consequence of a sale made through Nasdaq or any other recognised investment exchange (as defined in section 285 of the FSMA) or any other stock exchange on which the Company’s shares or depositary instruments representing such shares are normally traded; or |
| (iii) | a transfer which is shown to the satisfaction of the Board to be made in consequence of a sale of the whole of the beneficial interest in the shares to a person who is unconnected with the member and with any other person appearing to be interested in the shares. |
| 74.5 | Nothing contained in this Article shall be taken to limit the powers of the Company under section 794 of the Act. |
| 75. | Power of sale of shares of untraced members |
| 75.1 | The Company shall be entitled to sell at the best price reasonably obtainable any share of a member, or any share to which a person is entitled by transmission, if and provided that: |
| (a) | during the period of twelve years before the date of sending of the notice referred to in Article 75.1(b) no cheque, order or warrant in respect of such share sent by the Company through the post in a pre-paid envelope addressed to the member or to the person entitled by transmission to the share, at his or her address on the Register or other last known address given by the member or person to which cheques, orders or warrants in respect of such share are to be sent has been cashed and the Company has received no communications in respect of such share from such member or person entitled, provided that during such period of twelve years the Company has paid at least three cash dividends (whether interim or final) and no such dividend has been claimed by the person entitled to it; |
| (b) | on or after expiry of the said period of twelve years, the Company has given notice of its intention to sell such share by sending a notice to the member or person entitled by transmission to the share at his or her address on the Register or other last known address given by the member or person entitled by transmission to the share and before sending such a notice to the member or other person entitled by transmission, the Company must have used reasonable efforts to trace the member or other person entitled, engaging, if considered appropriate, a professional asset reunification company or other tracing agent and/or giving notice of its intention to sell the share by advertisement in a national newspaper and in a newspaper circulating in the area of the address of the member or person entitled by transmission to the share shown in the Register; |
| (c) | during the further period of three months following the date of such notice and prior to the exercise of the power of sale the Company has not received any communication in respect of such share from the member or person entitled by transmission; and |
| (d) | the Company has given notice to Nasdaq or the SEC of its intention to make such sale, if shares of the class concerned, or depositary instruments representing such shares, are listed on Nasdaq. |
| 75.2 | To give effect to any sale of shares under this Article: |
| (a) | in the case of a share in certificated form, the Board may authorise any person to execute an instrument of transfer of the share to the purchaser or a person nominated by the purchaser and take such other steps (including the giving of directions to or on behalf of the holder, who shall be bound by them) as it thinks fit to effect the transfer. The Board may authorise some person to transfer the shares in question and may enter the name of the transferee in respect of the transferred shares in the Register even if no share certificate has been lodged for such shares and may issue a new certificate to the transferee. An instrument of transfer executed by that person shall be as effective as if it had been executed by the holder of, or the person entitled by transmission to, the shares. |
| (b) | in the case of a share in uncertificated form, the Directors may: |
| (i) | to enable the Company to deal with the share in accordance with the provisions of this Article 75, require or procure any relevant person or the Operator (as applicable) to convert the share into certificated form; and |
| (ii) | after such conversion authorise any person to execute an instrument of transfer of the shares to the purchase or person nominated by the purchaser and take such other steps (including the giving of directions to or on behalf of the holder, who shall be bound by them) as it thinks fit to effect the transfer. |
| 75.3 | The buyer shall not be bound to see to the application of the purchase monies, nor shall his or her title to the shares be affected by any irregularity or invalidity in the proceedings in reference to the sale. If the shares are in uncertificated form, in accordance with the uncertificated securities rules, the Board may issue a written notification to the Operator requiring the conversion of the share to certificated form. |
| 75.4 | If during the period of twelve years referred to in Article 75.1, or during any period ending on the date when all the requirements of Articles 75.1(a) to 75.1(d) have been satisfied, any additional shares have been issued in respect of those held at the beginning of, or previously so issued during, any such period and all the requirements of Articles 75.1(b) to 75.1(d) have been satisfied in regard to such additional shares, the Company shall also be entitled to sell the additional shares. |
| 76. | Application of proceeds of sale of shares of untraced members |
The Company shall account to the member or other person entitled to the share for the net proceeds of a sale under Article 75 by carrying all monies relating to such sale to a separate account. The Company shall be deemed to be a debtor to, and not a trustee for, such member or other person in respect of such monies. Monies carried to such separate account may either be employed in the business of the Company or invested in such investments as the Board may think fit. No interest shall be payable to such member or other person in respect of such monies and the Company does not have to account for any money earned on them.
| 77. | Number of Directors |
Unless otherwise determined by the Company by ordinary resolution, the number of Directors (other than any alternate Directors) shall be at least two and not more than fifteen.
| 78. | Power of company to appoint Directors |
Subject to these Articles and the Companies Acts, the Company may by ordinary resolution appoint a person who is willing to act to be a Director, either to fill a vacancy or as an addition to the existing Board but the total number of Directors shall not exceed any maximum number fixed in accordance with these Articles.
| 79. | Power of Board to appoint Directors |
Subject to these Articles, the Board shall have power at any time to appoint any person who is willing to act as a Director, either to fill a vacancy or as an addition to the existing Board but the total number of Directors shall not exceed any maximum number fixed in accordance with these Articles.
| 80. | Eligibility of new Directors |
| 80.1 | No person, other than a retiring Director (by rotation or otherwise), shall be appointed or re-appointed a Director at any general meeting unless: |
| (a) | he or she is recommended by the Board; or |
| (b) | at least seven but not more than forty-two clear days before the date appointed for the meeting the Company has received notice from a member (other than the person proposed) entitled to vote at the meeting of his or her intention to propose a resolution for the appointment or re-appointment of that person, stating the particulars which would, if he or she were so appointed or re-appointed, be required to be included in the Company’s register of Directors and a notice executed by that person of his or her willingness to be appointed or re-appointed, is lodged at the Office. |
| 80.2 | A Director need not be a member of the Company. |
| 81. | Classes and Retirement of Directors |
| 81.1 | Following the Listing, the Directors shall be divided into three classes designated as “Class I”, “Class II” and “Class III”, respectively. The Board is authorised to assign (i) members of the Board already in office to such classes at the time the classification becomes effective and (ii) members of the Board who are so appointed following the Listing to such classes at the time of such appointment. |
| 81.2 | At the first annual general meeting of the Company following the Listing, each Director in Class I shall retire from office but shall be eligible for re-appointment by ordinary resolution at such annual general meeting and, in each case, where such Director is so re-appointed, they shall be entitled to serve until the third anniversary of such annual general meeting of the Company, at which stage such Director shall retire from office but shall be eligible for further re-appointment. |
| 81.3 | At the second annual general meeting of the Company following the Listing, each Director in Class II shall retire from office but shall be eligible for re-appointment by ordinary resolution at such annual general meeting and, in each case, where such Director is so re-appointed, they shall be entitled to serve until the third anniversary of such annual general meeting of the Company, at which stage such Director shall retire from office but shall be eligible for further re-appointment. |
| 81.4 | At the third annual general meeting of the Company following the Listing, each Director in Class III shall retire from office but shall be eligible for re-appointment by ordinary resolution at such annual general meeting and, in each case, where such Director is so re-appointed, they shall be entitled to serve until the third anniversary of such annual general meeting of the Company, at which stage such Director shall retire from office but shall be eligible for further re-appointment. |
| 81.5 | At each succeeding annual general meeting of the Company following the third annual general meeting of the Company after the Listing, Directors shall be elected to serve for a term of three years to succeed the Directors of the class whose terms expire at such annual general meeting. |
| 81.6 | Notwithstanding the foregoing provisions, each Director shall serve until their successor is duly elected and qualified or until their earlier death, resignation or removal. |
| 82. | Deemed re-appointment |
| 82.1 | A Director who retires at an annual general meeting shall (unless he or she is removed from office or his or her office is vacated in accordance with these Articles) retain office until the close of the meeting at which he or she retires or (if earlier) when a resolution is passed at that meeting not to fill the vacancy or to elect another person in his or her place or the resolution to re-appoint him or her is put to the meeting and lost. |
| 82.2 | If the Company, at any meeting at which a Director retires in accordance with these Articles does not fill the office vacated by such Director, the retiring Director, if willing to act, shall be deemed to be re-appointed unless at that meeting a resolution is passed not to fill the vacancy or elect another person in his or her place or unless the resolution to re-appoint him or her is put to the meeting and lost. |
| 83. | Procedure if insufficient Directors appointed |
| 83.1 | If: |
| (a) | at the annual general meeting in any year any resolution or resolutions for the appointment or re-appointment of the persons eligible for appointment or re-appointment as Directors are put to the meeting and lost; and |
| (b) | at the end of that meeting the number of Directors is fewer than any minimum number of Directors required under Article 77, |
all retiring Directors who stood for re-appointment at that meeting (Retiring Directors) shall be deemed to have been re-appointed as Directors and shall remain in office but the Retiring Directors may only act for the purpose of filling vacancies, convening general meetings of the Company and performing such duties as are essential to maintain the Company as a going concern, and not for any other purpose.
| 83.2 | The Retiring Directors shall convene a general meeting as soon as reasonably practicable following the meeting referred to in Article 83.1 and they shall retire from office at that meeting. If at the end of any meeting convened under this Article the number of Directors is fewer than any minimum number of Directors required under Article 77, the provisions of this Article shall also apply to that meeting. |
| 84. | Removal of Directors |
In addition to any power of removal conferred by the Companies Acts, the Company may by special resolution, or by ordinary resolution of which special notice has been given in accordance with section 312 of the Act, remove a Director before the expiry of his or her period of office (without prejudice to a claim for damages for breach of contract or otherwise) and may (subject to these Articles) by ordinary resolution appoint another person who is willing to act to be a Director in his or her place.
| 85. | Vacation of office by Director |
| 85.1 | Without prejudice to the provisions for retirement (by rotation or otherwise) contained in these Articles, the office of a Director shall be vacated if: |
| (a) | the Director resigns by notice in writing delivered to the Secretary at the Office or at an address specified by the Company for the purposes of communication by electronic means or tendered at a Board meeting; |
| (b) | the Director offers to resign by notice in writing delivered to the Secretary at the Office or at an address specified by the Company for the purposes of communication by electronic means or tendered at a Board meeting and the Board resolves to accept such offer; |
| (c) | the Director is requested to resign by all of the other Directors by notice in writing addressed to him or her at his or her address as shown in the register of Directors (without prejudice to any claim for damages which he or she may have for breach of any contract between him or her and the Company); |
| (d) | the Director ceases to be a Director by virtue of any provision of the Companies Acts, is removed from office pursuant to these Articles or the Act or becomes prohibited by law or by the rules of any applicable stock exchange from being a Director; |
| (e) | the Director becomes bankrupt or makes an arrangement or composition with his or her creditors generally; |
| (f) | a registered medical practitioner who is treating the Director gives a written opinion to the Company stating that he or she has become physically or mentally incapable of acting as a Director and may remain so for more than three months, or he or she is or has been suffering from mental or physical ill health and the Board resolves that his or her office be vacated; or |
| (g) | the Director is absent (whether or not any alternate Director appointed by him or her attends), without the permission of the Board, from Board meetings for six consecutive months and a notice is served on him or her personally, or at his or her residential address provided to the Company under section 165 of the Act signed by all the other Directors stating that he or she shall cease to be a Director with immediate effect (and such notice may consist of several copies each signed by one or more Directors). |
| 85.2 | If the office of a Director is vacated for any reason, he or she shall cease to be a member of any committee or sub-committee of the Board. |
| 86. | Resolution as to vacancy conclusive |
A resolution of the Board declaring a Director to have vacated office under the terms of Article 85 shall be conclusive as to the fact and ground of vacation stated in the resolution.
| 87. | Appointment of alternate Directors |
| 87.1 | Each Director may appoint any person (including another Director) to be his or her alternate and may at his or her discretion remove an alternate Director so appointed. Any appointment or removal of an alternate Director must be by written notice delivered to the Office or at an address specified by the Company for the purposes of communication by electronic means or tendered at a Board meeting or in any other manner approved by the Board. The appointment requires the approval of the Board unless it has been previously approved or the appointee is another Director. |
| 87.2 | An alternate Director must provide the particulars, and sign any form for public filing required by the Companies Acts relating to his or her appointment. |
| 88. | Alternate Directors’ participation in Board meetings |
| 88.1 | Every alternate Director is (subject to his or her giving to the Company an address within the United Kingdom at which notices may be served on him or her (and, if applicable, an address in relation to which electronic communications may be received by him or her)) entitled to receive notice of all meetings of the Board and all committees of the Board of which his or her appointor is a member and, in his or her appointor’s absence, to attend and vote at such meetings and to exercise all the powers, rights, duties and authorities of his or her appointor. Each person acting as an alternate Director shall have a separate vote at Board meetings for each Director for whom he or she acts as alternate Director in addition to his or her own vote if he or she is also a Director, but he or she shall count as only one for the purpose of determining whether a quorum is present. |
| 88.2 | Signature by an alternate Director of any resolution in writing of the Board or a committee of the Board will, unless the notice of his or her appointment provides otherwise, be as effective as signature by his or her appointor. |
| 89. | Alternate Director responsible for own acts |
Each person acting as an alternate Director will be an officer of the Company, will alone be responsible to the Company for his or her own acts and defaults and will not be deemed to be the agent of the Director appointing him or her.
| 90. | Interests of alternate Director |
An alternate Director is entitled to contract and be interested in and benefit from contracts or arrangements with the Company, to be repaid expenses and to be indemnified to the same extent as if he or she were a Director. However, he or she is not entitled to receive from the Company any fees for his or her services as alternate, except such part (if any) of the fee payable to his or her appointor as such appointor may by written notice to the Company direct.
| 91. | Revocation of alternate Director |
An alternate Director will cease to be an alternate Director:
| (a) | if his or her appointor revokes his or her appointment; or |
| (b) | if he or she resigns his or her office by notice in writing to the Company; or |
| (c) | if his or her appointor ceases for any reason to be a Director, provided that if any Director retires but is re-appointed or deemed to be re-appointed at the same meeting, any valid appointment of an alternate Director which was in force immediately before his or her retirement shall remain in force; or |
| (d) | if any event happens in relation to him or her which, if he or she were a Director otherwise appointed, would cause him or her to vacate his or her office. |
| 92. | Directors’ fees |
Each of the Directors may be paid a fee at such rate as may from time to time be determined by the Board. However, the aggregate of all fees payable to the Directors (other than amounts payable under any other provision of these Articles) must not exceed $2,500,000 a year or such higher amount as may from time to time be decided by ordinary resolution of the Company. Any fees payable under this Article shall be distinct from any salary, remuneration or other amounts payable to a Director under any other provisions of these Articles and shall accrue from day to day.
| 93. | Expenses |
Each Director may be paid his or her reasonable travelling, hotel and other expenses properly incurred by him or her in or about the performance of his or her duties as Director, including any expenses incurred in attending meetings of the Board or any committee of the Board or general meetings or separate meetings of the holders of any class of shares or debentures of the Company. Subject to the Act, the Directors shall have the power to make arrangements to provide a Director with funds to meet expenditure incurred or to be incurred by him or her for the purposes of the Company or for the purpose of enabling him or her to perform his or her duties as an officer of the Company or to enable him or her to avoid incurring any such expenditure.
| 94. | Additional remuneration |
If by arrangement with the Board any Director shall perform or render any special duties or services outside his or her ordinary duties as a Director and not in his or her capacity as a holder of employment or executive office, he or she may be paid such reasonable additional remuneration (whether by way of salary, commission, participation in profits or otherwise) as the Board may determine.
| 95. | Remuneration of executive Directors |
The salary or remuneration of any Director appointed to hold any employment or executive office in accordance with these Articles may be either a fixed sum of money, or may altogether or in part be governed by business done or profits made or otherwise determined by the Board, and may be in addition to or instead of any fee payable to him or her for his or her services as Director under these Articles.
| 96. | Pensions and other benefits |
| 96.1 | The Board may exercise all the powers of the Company to provide pensions or other retirement or superannuation benefits and to provide death or disability benefits or other allowances or gratuities (whether by insurance or otherwise) for any person who is or has at any time been a Director or employee of: |
| (a) | the Company; |
| (b) | any company which is or was a holding company or a subsidiary undertaking of the Company; |
| (c) | any company which is or was allied to or associated with the Company or a subsidiary undertaking or holding company of the Company; or |
| (d) | a predecessor in business of the Company or of any holding company or subsidiary undertaking of the Company, |
and, in each case, for any member of his or her family (including a spouse or former spouse) and any person who is or was dependent on him or her.
| 96.2 | The Board may establish, maintain, subscribe and contribute to any scheme, institution, association, club, trust or fund and pay premiums and, subject to the Companies Acts, lend money or make payments to, guarantee or give an indemnity in respect of, or give any financial or other assistance in connection with any of the matters set out in Article 96.1 above. The Board may procure any of such matters to be done by the Company either alone or in conjunction with any other person. Any Director or former Director shall be entitled to receive and retain for his or her own benefit any pension or other benefit provided under this Article and shall not have to account for it to the Company. The receipt of any such benefit will not disqualify any person from being or becoming a Director of the Company. |
| 97. | Powers of the Board |
| 97.1 | Subject to the Companies Acts, these Articles and to any directions given by special resolution of the Company, the business of the Company will be managed by the Board, which may exercise all the powers of the Company, whether relating to the management of the business or not. |
| 97.2 | No alteration of these Articles and no such direction given by the Company shall invalidate any prior act of the Board which would have been valid if such alteration had not been made or such direction had not been given. Provisions contained elsewhere in these Articles as to any specific power of the Board shall not be deemed to limit the general powers given by this Article. |
| 98. | Powers of Directors if less than minimum number |
If the number of Directors is less than the minimum prescribed in Article 77 or decided by the Company by ordinary resolution, the remaining Director or Directors may act only for the purposes of appointing an additional Director or Directors to make up that minimum or convening a general meeting of the Company for the purpose of making such appointment. If no Director or Directors is or are able or willing to act, two members may convene a general meeting for the purpose of appointing Directors. An additional Director appointed in this way holds office (subject to these Articles) only until the dissolution of the next annual general meeting after his or her appointment unless he or she is reappointed during the annual general meeting.
| 99. | Powers of executive Directors |
The Board or any committee authorised by the Board may:
| (a) | delegate or entrust to and confer on any Director holding executive office (including a Chief Executive or Managing Director) such of its powers, authorities and discretions (with power to sub-delegate) for such time, on such terms and subject to such conditions as it thinks fit; and |
| (b) | revoke, withdraw, alter or vary all or any of such powers. |
| 100. | Delegation to committees |
| 100.1 | The Board may delegate any of its powers, authorities and discretions (with power to sub-delegate) for such time on such terms and subject to such conditions as it thinks fit to any committee consisting of one or more Directors and (if thought fit) one or more other persons provided that: |
| (a) | a majority of the members of a committee shall be Directors; and |
| (b) | no resolution of a committee shall be effective unless a majority of those present when it is passed are Directors or alternate Directors. |
| 100.2 | The Board may confer such powers either collaterally with, or to the exclusion of and in substitution for, all or any of the powers of the Board in that respect and may revoke, withdraw, alter or vary any such powers and discharge any such committee in whole or in part. Insofar as any power, authority or discretion is so delegated, any reference in these Articles to the exercise by the Board of such power, authority or discretion shall be construed as if it were a reference to the exercise of such power, authority or discretion by such committee. |
| 101. | Local management |
| 101.1 | The Board may establish any local or divisional boards or agencies for managing any of the affairs of the Company in any specified locality, either in the United Kingdom or elsewhere, and appoint any persons to be members of such local or divisional board, or any managers or agents, and may fix their remuneration. |
| 101.2 | The Board may delegate to any local or divisional board, manager or agent so appointed any of its powers, authorities and discretions (with power to sub-delegate) and may authorise the members of any such local or divisional board, or any of them, to fill any vacancies and to act notwithstanding vacancies. Any such appointment or delegation under this Article 101 may be made, on such terms and conditions as the Board may think fit. The Board may confer such powers either collaterally with, or to the exclusion of and in substitution for, all or any of the powers of the Board, and the Board may remove any person so appointed and may annul or vary all or any of such powers, but no person dealing in good faith and without notice of any such annulment or variation shall be affected by it. |
| 101.3 | Subject to any terms and conditions expressly imposed by the Board, the proceedings of any local or divisional board or agency with two or more members shall be governed by such of these Articles as regulate the proceedings of the Board, so far as they are capable of applying. |
| 102. | Power of attorney |
The Board may, by power of attorney or otherwise, appoint any person or persons to be the agent or attorney of the Company and may delegate to any such person or persons any of its powers, authorities and discretions (with power to sub-delegate), in each case for such purposes and for such time, on such terms (including as to remuneration) and conditions as it thinks fit. The Board may confer such powers either collaterally with, or to the exclusion of and in substitution for, all or any of the powers of the Board in that respect and may revoke, withdraw, alter or vary any of such powers.
| 103. | Exercise of voting power |
The Board may exercise or cause to be exercised the voting power conferred by the shares in any other company held or owned by the Company, or any power of appointment to be exercised by the Company, in such manner as it thinks fit (including the exercise of the voting power or power of appointment in favour of the appointment of any Director as a Director or other officer or employee of such company or in favour of the payment of remuneration to the Directors, officers or employees of such company).
| 104. | Provision for employees on cessation of business |
The Board may, by resolution, sanction the exercise of the power to make provision for the benefit of persons employed or formerly employed by the Company or any of its subsidiary undertakings, in connection with the cessation or the transfer to any person of the whole or part of the undertaking of the Company or that subsidiary undertaking, but any such resolution shall not be sufficient for payments to or for the benefit of Directors, former Directors or shadow Directors.
| 105. | Overseas registers |
Subject to the Companies Acts, the Company may keep an overseas, local or other register and the Board may make and vary such regulations as it thinks fit respecting the keeping of any such register.
| 106. | Borrowing powers |
Subject to these Articles and the Companies Acts, the Board may exercise all the powers of the Company to:
| (a) | borrow money; |
| (b) | indemnify and guarantee; |
| (c) | mortgage or charge all or any part of the undertaking, property and assets (present and future) and uncalled capital of the Company; |
| (d) | create and issue debentures and other securities; and |
| (e) | give security either outright or as collateral security for any debt, liability or obligation of the Company or of any third party. |
| 107. | Board meetings |
| 107.1 | The Board can decide when and where to have meetings and how they will be conducted. They may also adjourn meetings. |
| 107.2 | A Board meeting can be called by any Director. The Secretary must call a Board meeting if asked to do so by a Director. |
| 108. | Notice of Board meetings |
| 108.1 | Notice of a Board meeting shall be deemed to be duly given to a Director if it is given to him or her personally or by word of mouth or given in writing or by electronic means to him or her at his or her last known address or any other address given by him or her to the Company for that purpose. |
| 108.2 | A Director may waive the requirement that notice be given to him or her of any Board meeting, either prospectively or retrospectively and any retrospective waiver shall not affect the validity of the meeting or of any business conducted at the meeting. |
| 109. | Quorum |
| 109.1 | The quorum necessary for the transaction of business shall be at least two persons, each being a Director or an alternate Director. A duly convened meeting of the Board at which a quorum is present shall be competent to exercise all or any of the authorities, powers, and discretions for the time being vested in or exercisable by the Board. |
| 109.2 | If a Director ceases to be a Director at a Board meeting, he or she can continue to be present and to act as a Director and be counted in the quorum until the end of the meeting if no other Director objects and if otherwise a quorum of Directors would not be present. |
| 110. | Chair |
| 110.1 | The Board may appoint one or more of its body as chair or joint chair and one or more of its body as deputy chair of its meetings and may determine the period for which he or she is or they are to hold office and may at any time remove him or her or them from office. |
| 110.2 | If no such chair or deputy chair is elected, or if at any meeting neither a chair nor a deputy chair is present within ten minutes of the time appointed for holding the same, the Directors present shall choose one of their number to be chair of such meeting. In the event two or more joint chairmen or, in the absence of a chair, two or more deputy chairs being present, the joint chair or deputy chair to act as chair of the meeting shall be decided by those Directors present. |
| 111. | Voting |
Questions arising at any Board meeting shall be determined by a majority of votes. In the case of an equality of votes the chair of that meeting shall have a second or casting vote (unless he or she is not entitled to vote on the resolution in question).
| 112. | Participation by telephone or other form of communication |
| 112.1 | Any Director or his or her alternate may validly participate in a meeting of the Board or a committee of the Board through the medium of conference telephone or any other form of communications equipment (whether in use when these Articles are adopted or developed subsequently), provided that all persons participating in the meeting are able to hear and speak to each other throughout such meeting. |
| 112.2 | A person so participating by telephone or other communication shall be deemed to be present in person at the meeting and shall be counted in a quorum and entitled to vote. |
| 112.3 | A resolution passed at any meeting held in the above manner, and signed by the chair of the meeting, shall be as valid and effectual as if it had been passed at a meeting of the Board (or committee, as the case may be) duly convened and held. |
| 113. | Resolution in writing |
| 113.1 | A resolution in writing signed or confirmed electronically by all the Directors for the time being entitled to receive notice of a Board meeting and to vote on the resolution and not being less than a quorum (or by all the members of a committee of the Board for the time being entitled to receive notice of such committee meeting and to vote on the resolution and not being less than a quorum of that committee), shall be as valid and effective for all purposes as a resolution duly passed at a meeting of the Board (or committee, as the case may be). |
| 113.2 | Such a resolution may consist of several documents or electronic communications in the same form each signed or authenticated by one or more of the Directors or members of the relevant committee. |
| 114. | Proceedings of committees |
All committees of the Board shall, in the exercise of the powers delegated to them and in the transaction of business, conform with any mode of proceedings and regulations which the Board may prescribe and subject to this shall be governed by such of these Articles as regulate the proceedings of the Board as are capable of applying.
| 115. | Minutes of proceedings |
| 115.1 | The Board shall keep minutes of all meetings of members, all Board meetings and meetings of committees of the Board. The minutes must include the names of the Directors present. |
| 115.2 | Any such minutes, if purporting to be signed by the chair of the meeting at which the proceedings were held or by the chair of the next meeting or the Secretary, shall be evidence of the matters stated in such minutes without any further proof. |
| 116. | Validity of proceedings |
All acts done by a meeting of the Board, or of a committee of the Board, or by any person acting as a Director, alternate Director or member of a committee shall be valid even if it is discovered afterwards that there was some defect in the appointment of any person or persons acting, or that they or any of them were or was disqualified from holding office or not entitled to vote, or had in any way vacated their or his or her office.
| 117. | Transactions or other arrangements with the company |
| 117.1 | Subject to the Companies Acts and provided he or she has declared the nature and extent of his or her interest in accordance with the requirements of the Companies Acts, a Director who is in any way, whether directly or indirectly, interested in an existing or proposed transaction or arrangement with the Company may: |
| (a) | be a party to, or otherwise interested in, any transaction or arrangement with the Company or in which the Company is otherwise (directly or indirectly) interested; |
| (b) | act by himself or herself or through his or her firm in a professional capacity for the Company (otherwise than as auditor) and he or she or his or her firm shall be entitled to remuneration for professional services as if he or she were not a Director; |
| (c) | be or become a Director or other officer of, or employed by, or a party to a transaction or arrangement with, or otherwise interested in, any body corporate in which the Company is otherwise (directly or indirectly) interested; and |
| (d) | hold any office or place of profit with the Company (except as auditor) in conjunction with his or her office of Director for such period and upon such terms, including as to remuneration as the Board may decide. |
| 117.2 | A Director shall not, save as he or she may otherwise agree, be accountable to the Company for any benefit which he or she derives from any such contract, transaction or arrangement or from any such office or employment or from any interest in any such body corporate and no such contract, transaction or arrangement shall be liable to be avoided on the grounds of any such interest or benefit nor shall the receipt of any such remuneration or other benefit constitute a breach of his or her duty under section 176 of the Act. |
| 118. | Authorisation of Directors’ conflicts of interest |
| 118.1 | The Board may, in accordance with the requirements set out in this Article, authorise any matter or situation proposed to them by any Director which would, if not authorised, involve a Director (an Interested Director) breaching his or her duty under the Act to avoid conflicts of interest. |
| 118.2 | A Director seeking authorisation in respect of a conflict of interest shall declare to the Board the nature and extent of his or her interest in a conflict of interest as soon as is reasonably practicable. The Director shall provide the Board with such details of the matter as are necessary for the Board to decide how to address the conflict of interest together with such additional information as may be requested by the Board. |
| 118.3 | Any authorisation under this Article will be effective only if: |
| (a) | to the extent permitted by the Act, the matter in question shall have been proposed by any Director for consideration in the same way that any other matter may be proposed to the Directors under the provisions of these Articles; |
| (b) | any requirement as to the quorum for consideration of the relevant matter is met without counting the Interested Director and any other interested Director; and |
| (c) | the matter is agreed to without the Interested Director voting or would be agreed to if the Interested Director’s and any other interested Director’s vote is not counted. |
| 118.4 | Any authorisation of a conflict of interest under this Article must be recorded in writing (but the authority shall be effective whether or not the terms are so recorded) and may (whether at the time of giving the authorisation or subsequently): |
| (a) | extend to any actual or potential conflict of interest which may reasonably be expected to arise out of the matter or situation so authorised; |
| (b) | provide that the Interested Director be excluded from the receipt of documents and information and the participation in discussions (whether at meetings of the Directors or otherwise) related to the conflict of interest; |
| (c) | impose upon the Interested Director such other terms for the purposes of dealing with the conflict of interest as the Directors think fit; |
| (d) | provide that, where the Interested Director obtains, or has obtained (through his or her involvement in the conflict of interest and otherwise than through his or her position as a Director) information that is confidential to a third party, he or she will not be obliged to disclose that information to the Company, or to use it in relation to the Company’s affairs where to do so would amount to a breach of that confidence; and |
| (e) | permit the Interested Director to absent himself or herself from the discussion of matters relating to the conflict of interest at any meeting of the Directors and be excused from reviewing papers prepared by, or for, the Directors to the extent they relate to such matters. |
| 118.5 | Where the Directors authorise a conflict of interest, the Interested Director will be obliged to conduct himself or herself in accordance with any terms and conditions imposed by the Directors in relation to the conflict of interest. |
| 118.6 | The Directors may revoke or vary such authorisation at any time, but this will not affect anything done by the Interested Director, prior to such revocation or variation, in accordance with the terms of such authorisation. |
| 118.7 | A Director is not required, by reason of being a Director (or because of the fiduciary relationship established by reason of being a Director), to account to the Company for any remuneration, profit or other benefit which he or she derives from or in connection with a relationship involving a conflict of interest which has been authorised by the Directors or by the Company in general meeting (subject in each case to any terms, limits or conditions attaching to that authorisation) and no contract shall be liable to be avoided on such grounds. |
| 118.8 | If he or she has disclosed to the Board the nature and extent of his or her interest to the extent required by the Companies Acts, a Director is not required, by reason of being a Director (or because of the fiduciary relationship established by reason of being a Director), to account to the Company for any remuneration or other benefit which he or she derives from or in connection with: |
| (a) | being a party to, or otherwise interested in, any transaction or arrangement with: |
| (i) | the Company or in which the Company is interested; or |
| (ii) | a body corporate in which the Company is interested; |
| (b) | acting (otherwise than as auditor) alone or through his or her organisation in a professional capacity for the Company (and he or she or that organisation is entitled to remuneration for professional services as if he or she were not a Director); or |
| (c) | being a director or other officer of, or employed by, or otherwise interested in any other body corporate in which the Company is interested. |
| 118.9 | A Director’s receipt of any remuneration or other benefit referred to in Articles 118.7 or 118.8 does not constitute an infringement of his or her duty under the Companies Acts. |
| 118.10 | A transaction or arrangement referred to in Articles 118.7 or 118.8 is not liable to be avoided on the ground of any remuneration, benefit or interest referred to in that Article. |
| 119. | Directors’ permitted interests |
| 119.1 | A Director cannot vote or be counted in the quorum on any resolution relating to any transaction or arrangement with the Company in which he or she has an interest and which may reasonably be regarded as likely to give rise to a conflict of interest but can vote (and be counted in the quorum) on the following: |
| (a) | giving him or her any security, guarantee or indemnity for any money or any liability which he, she or any other person, has lent or obligations he or she or any other person has undertaken at the request, or for the benefit, of the Company or any of its subsidiary undertakings; |
| (b) | giving any security, guarantee or indemnity to any other person for a debt or obligation which is owed by the Company or any of its subsidiary undertakings, to that other person if the Director has taken responsibility for some or all of that debt or obligation. The Director can take this responsibility by giving a guarantee, indemnity or security; |
| (c) | a proposal or contract relating to an offer of any shares or debentures or other securities for subscription or purchase by the Company or any of its subsidiary undertakings, if the Director takes part because he or she is a holder of shares, debentures or other securities, or if he or she takes part in the underwriting or sub-underwriting of the offer; |
| (d) | any arrangement for the benefit of employees of the Company or any of its subsidiary undertakings which only gives him or her benefits which are also generally given to employees to whom the arrangement relates; |
| (e) | any arrangement involving any other company if the Director (together with any person connected with the Director) has an interest of any kind in that company (including an interest by holding any position in that company or by being a member of that company). This does not apply if he or she knows that he or she has a Relevant Interest; |
| (f) | a contract relating to insurance which the Company can buy or renew for the benefit of the Directors or a group of people which includes Directors; and |
| (g) | a contract relating to a pension, superannuation or similar scheme or a retirement, death, disability benefits scheme or employees’ share scheme which gives the Director benefits which are also generally given to the employees to whom the scheme relates. |
| 119.2 | A Director cannot vote or be counted in the quorum on a resolution relating to his or her own appointment or the settlement or variation of the terms of his or her appointment to an office or place of profit with the Company or any other company in which the Company has an interest. |
| 119.3 | Where the Directors are considering proposals about the appointment, or the settlement or variation of the terms or the termination of the appointment of two or more Directors to other offices or places of profit with the Company or any company in which the Company has an interest, a separate resolution may be put in relation to each Director and in that case each of the Directors concerned shall be entitled to vote and be counted in the quorum in respect of each resolution unless it concerns his or her own appointment or the settlement or variation of the terms or the termination of his or her own appointment or the appointment of another Director to an office or place of profit with a company in which the Company has an interest and the Director seeking to vote or be counted in the quorum has a Relevant Interest in it. |
| 119.4 | A company shall be deemed to be one in which the Director has a Relevant Interest if and so long as (but only if and so long as) he or she is to his or her knowledge (either directly or indirectly) the holder of or beneficially interested in one per cent. or more of any class of the equity share capital of that company (calculated exclusive of any shares of that class in that company held as treasury shares) or of the voting rights available to members of that company. In relation to an alternate Director, an interest of his or her appointor shall be treated as an interest of the alternate Director without prejudice to any interest which the alternate Director has otherwise. Where a company in which a Director has a Relevant Interest is interested in a contract, he or she also shall be deemed interested in that contract. |
| 119.5 | If a question arises at a Board meeting about whether a Director (other than the chair of the meeting) has an interest which is likely to give rise to a conflict of interest, or whether he or she can vote or be counted in the quorum, and the Director does not agree to abstain from voting on the issue or not to be counted in the quorum, the question must be referred to the chair of the meeting. The chair’s ruling about the relevant Director is final and conclusive, unless the nature and extent of the Director’s interests have not been fairly disclosed to the Directors. If the question arises about the chair of the meeting, the question must be directed to the Directors. The chair cannot vote on the question but can be counted in the quorum. The Directors’ resolution about the chair is final and conclusive, unless the nature and extent of the chair’s interests have not been fairly disclosed to the Directors. |
| 120. | General |
For the purposes of Articles 117 to 119 inclusive (which shall apply equally to alternate Directors):
| 120.1 | An interest of a person who is connected (which word shall have the meaning given to it by section 252 of the Act) with a Director shall be treated as an interest of the Director. |
| 120.2 | A contract includes references to any proposed contract and to any transaction or arrangement or proposed transaction or arrangement whether or not constituting a contract. |
| 120.3 | A conflict of interest includes a conflict of interest and duty and a conflict of duties. |
| 120.4 | Subject to the Companies Acts, the Company may by ordinary resolution suspend or relax the provisions of Articles 117 to 119 to any extent or ratify any contract not properly authorised by reason of a contravention of any of the provisions of Articles 117 to 119. |
| 121. | Power to authenticate documents |
Any Director, the Secretary or any person appointed by the Board for the purpose shall have power to authenticate any documents affecting the constitution of the Company and any resolution passed by the Company or the Board or any committee, and any books, records, documents and accounts relating to the business of the Company, and to certify copies or extracts as true copies or extracts. Where any books, records, documents or accounts are not at the Office, the local manager or other officer of the Company who has their custody shall be deemed to be a person appointed by the Board for this purpose. A document purporting to be a copy of a resolution, or an extract from the minutes of a meeting, of the Company or the Board or any committee which is so certified shall be conclusive evidence in favour of all persons dealing with the Company that such resolution has been duly passed or, as the case may be, that any minute so extracted is a true and accurate record of proceedings at a duly constituted meeting.
| 122. | Use of seals |
| 122.1 | The Board shall provide for the safe custody of the Seal. A Seal shall not be used without the authority of the Board or of a committee of the Board so authorised. |
| 122.2 | Subject as otherwise provided in these Articles, every document which is sealed using the Seal must be signed by at least one authorised person in the presence of a witness who attests the signature. An authorised person for this purpose is any Director, the Secretary or any other person authorised by the Directors for the purpose of signing documents to which the Seal is applied. |
| 122.3 | The Seal shall be used only for sealing securities issued by the Company and documents creating or evidencing securities so issued. Any such securities or documents sealed with the Seal shall not require to be signed unless the Board decides otherwise or the law otherwise requires. |
| 122.4 | The Board may decide who will sign an instrument to which a Seal is affixed (or in the case of a share certificate, on which the Seal may be printed) either generally or in relation to a particular instrument or type of instrument and may also determine either generally or in a particular case that a signature may be dispensed with or affixed by mechanical means. |
| 123. | Declaration of dividends |
Subject to the Act and these Articles, the Company may by ordinary resolution declare dividends to be paid to members according to their respective rights and interests in the profits of the Company. However, no dividend shall exceed the amount recommended by the Board.
| 124. | Interim dividends |
| 124.1 | Subject to the Act, the Board may declare and pay such interim dividends (including any dividend at a fixed rate) as appears to the Board to be justified by the profits of the Company available for distribution. If the Board acts in good faith, it shall not incur any liability to the holders of shares for any loss that they may suffer by the lawful payment of any interim dividend on any other class of shares ranking with or after those shares. |
| 124.2 | If the share capital is divided into different classes, the Board may pay interim dividends on shares which confer deferred or non-preferred rights with regard to dividends as well as on shares which confer preferential rights with regard to dividends, but no interim dividend shall be paid on shares carrying deferred or non-preferred rights if, at the time of payment, any preferential dividend is in arrears. |
| 124.3 | The Board may also pay at intervals settled by them any dividend payable at a fixed rate if it appears to them that the profits available for distribution justify the payment. If the Directors act in good faith they shall not incur any liability to the holders of shares conferring preferred rights for any loss they may suffer by the lawful payment of a dividend on any shares having deferred or non-preferred rights. |
| 125. | Calculation and currency of dividends |
Except as provided otherwise by the rights attached to shares, all dividends:
| (a) | shall be declared and paid according to the amounts paid up (otherwise than in advance of calls) on the shares on which the dividend is paid; |
| (b) | shall be apportioned and paid proportionately to the amounts paid up on the shares during any portion or portions of the period in respect of which the dividend is paid, but if any share is issued on terms that it shall rank for dividend as from a particular date, it shall rank for dividend accordingly; and |
| (c) | may be declared or paid in any currency. The Board may decide the rate of exchange for any currency conversions that may be required and how any costs involved are to be met. |
| 126. | Amounts due on shares can be deducted from dividends |
The Board may deduct from any dividend or other money payable to any person on or in respect of a share all such sums as may be due from him or her to the Company on account of calls or otherwise in relation to the shares of the Company. Sums so deducted can be used to pay amounts owing to the Company in respect of the shares.
| 127. | Dividends not in cash |
The Board may, by ordinary resolution of the Company direct, or in the case of an interim dividend may without the authority of an ordinary resolution direct, that payment of any dividend declared may be satisfied wholly or partly by the distribution of assets, and in particular of paid up shares or debentures of any other company, or in any one or more of such ways. Where any difficulty arises regarding such distribution, the Board may settle it as it thinks fit. In particular, the Board may:
| (a) | issue fractional certificates (or ignore fractions); |
| (b) | fix the value for distribution of such assets or any part of them and determine that cash payments may be made to any members on the footing of the values so fixed, in order to adjust the rights of members; and |
| (c) | vest any such assets in trustees on trust for the person entitled to the dividend. |
| 128. | No interest on dividends |
Unless otherwise provided by the rights attached to the share, no dividend or other monies payable by the Company or in respect of a share shall bear interest as against the Company.
| 129. | Method of payment |
| 129.1 | The Company may pay any dividend, interest or other sum payable in respect of a share wholly or partly in cash or by direct debit, bank transfer, cheque, dividend warrant, or money order or by any other method, including by electronic means, as the Board may consider appropriate. For uncertificated shares, any payment may be made by means of the relevant system (subject always to the facilities and requirements of the relevant system) and such payment may be made by the Company or any person on its behalf by sending an instruction to the operator of the relevant system to credit the cash memorandum account of the holder or joint holders of such shares or, if permitted by the Company, of such person as the holder or joint holders may in writing direct. |
| 129.2 | The Company may send such payment by post or other delivery service (or by such means offered by the Company as the member or person entitled to it may agree in writing) to the registered address of the member or person entitled to it (or, if two or more persons are holders of the share or are jointly entitled to it because of the death or bankruptcy of the member or otherwise by operation of law, to the registered address of such of those persons as is first named in the Register) or to such person and such address as such member or person may direct in writing. |
| 129.3 | Every cheque, warrant, order or other form of payment is sent at the risk of the person entitled to the money represented by it, shall be made payable to the person or persons entitled, or to such other person as the person or persons entitled may direct in writing. Payment of the cheque, warrant, order or other form of payment (including transmission of funds through a bank transfer or other funds transfer system or by such other electronic means as permitted by these Articles or in accordance with the facilities and requirements of the relevant system concerned) shall be good discharge to the Company. If any such cheque, warrant, order or other form of payment has or shall be alleged to have been lost, stolen or destroyed the Company shall not be responsible. |
| 129.4 | Any joint holder or other person jointly entitled to a share may give an effective receipt for any dividend or other monies payable in respect of such share. |
| 129.5 | The Board may, at its discretion, make provisions to enable any member as the Board shall determine to receive duly declared dividends in a currency or currencies other than sterling. For the purposes of the calculation of the amount receivable in respect of any dividend, the rate of exchange to be used to determine the foreign currency equivalent of any sum payable as a dividend shall be such rate or rates and the payment shall be on such terms and conditions as the Board may in its absolute discretion determine. |
| 129.6 | In respect of the payment of any dividend or other sum which is a distribution, the Board may decide, and notify recipients, that: |
| (a) | one or more of the means described in this Article 129 will be used for payment and a recipient may elect to receive the payment by one of the means so notified in the manner prescribed by the Directors; |
| (b) | one or more of such means will be used for the payment unless a recipient elects otherwise in the manner prescribed by the Directors; or |
| (c) | one or more of such means will be used for the payment and that recipients will not be able to elect otherwise, |
the Board may for this purpose decide that different methods of payment may apply to different recipients or groups of recipients.
| 129.7 | All cheques, warrants and similar financial instruments are sent, and payment in any other way is made, at the risk of the person who is entitled to the money and the Company will not be responsible for a payment which is lost, rejected or delayed. The Company can rely on a receipt for a dividend or other money paid in relation to a share from any one of the joint recipients on behalf of all of them. The Company is treated as having paid a dividend if the cheque, warrant or similar financial instrument is cleared or if a payment is made using a relevant system or inter-bank transfer or other electronic means. |
| 129.8 | Subject to the rights attaching to any shares, any dividends or other monies payable on or in respect of a share may be declared or paid in such currency or currencies and using such exchange rate or such date for determining the value or currency conversions as the Directors may determine. |
| 130. | Uncashed dividends |
If cheques, warrants or orders for dividends or other sums payable in respect of a share sent by the Company to the person entitled to them are returned to the Company or left uncashed on two consecutive occasions or, following one occasion, reasonable enquires have failed to establish any new address to be used for the purpose, the Company does not have to send any dividends or other monies payable in respect of that share due to that person until he or she notifies the Company of an address to be used for the purpose. If any such cheque, warrant or order has or is alleged to have been lost, stolen or destroyed, the Directors may, on request of the person entitled to it, issue a replacement cheque, warrant or order subject to compliance with such conditions as to evidence and indemnity and the payment of out of pocket expenses of the Company in connection with the request as the Directors may think fit.
| 131. | Unclaimed dividends |
All dividends, interest or other sums payable and unclaimed for twelve months after having become payable may be invested or otherwise made use of by the Board for the benefit of the Company until claimed. The Company shall not be a trustee in respect of such unclaimed dividends and will not be liable to pay interest on it. All dividends that remain unclaimed for twelve years after they were first declared or became due for payment shall (if the Board so resolves) be forfeited and shall cease to remain owing by the Company.
| 132. | Scrip dividends |
| 132.1 | Subject to the Act, the Board may, by ordinary resolution of the Company and subject to such terms and conditions as the Board may determine, offer to any holders of shares (excluding any member holding shares as treasury shares) the right to elect to be issued with shares, credited as fully paid, instead of cash in respect of the whole (or some part, to be determined by the Board) of any dividend specified by the ordinary resolution. The following provisions shall apply: |
| (a) | the said resolution may specify a particular dividend, or may specify all or any dividends declared within a specified period or periods but such period may not end later than the fifth anniversary of the date of the meeting at which the ordinary resolution is passed; |
| (b) | the entitlement of each holder of shares to new shares shall be such that the relevant value of the entitlement shall be as nearly as possible equal to (but not greater than) the cash amount (disregarding any tax credit) of the dividend that such holder would have received by way of dividend. For this purpose relevant value shall be calculated by reference to the average of the middle market quotations for the shares or depositary instruments representing such shares, on Nasdaq (or any other publication of a recognised investment exchange showing quotations for the Company’s shares), for the day on which the shares are first quoted “ex” the relevant dividend and the four subsequent dealing days, or in such other manner as the Board may determine on such basis as it considers to be fair and reasonable. A certificate or report by the Company’s auditors as to the amount of the relevant value in respect of any dividend shall be conclusive evidence of that amount; |
| (c) | no fractions of a share shall be allotted. The Board may make such provisions as it thinks fit for any fractional entitlements including provisions where, in whole or in part, the benefit accrues to the Company and/or under which fractional entitlements are accrued and/or retained and in each case accumulated on behalf of any member and such accruals or retentions are applied to the allotment by way of bonus to or cash subscription on behalf of any member of fully paid shares and/or provisions where cash payments may be made to members in respect of their fractional entitlements; |
| (d) | the Board shall, after determining the basis of allotment, notify the holders of shares in writing of the right of election offered to them, and specify the procedure to be followed and place at which, and the latest time by which, elections must be lodged in order to be effective. No such notice need to be given to holders of shares who have previously given election mandates in accordance with this Article and whose mandates have not been revoked. The accidental omission to give notice of any right of election to, or the non-receipt (even if the Company becomes aware of such non-receipt) of any such notice by, any holder of shares entitled to the same shall neither invalidate any offer of an election nor give rise to any claim, suit or action; |
| (e) | the Board may on any occasion decide that rights of election shall only be made available subject to such exclusions, restrictions or other arrangements as they shall in their absolute discretion deem necessary or desirable in order to comply with legal or practical problems under the laws of, or the requirements of any recognised regulatory body or stock exchange in, any territory; |
| (f) | the Board shall not proceed with any election unless the company has sufficient reserves or funds that may be capitalised, and the Board has authority to allot sufficient shares, to give effect to it after the basis of the allotment is determined; |
| (g) | the Board may exclude from any offer or make other arrangements in relation to any holders of shares where the Board considers that the making of the offer to them or in respect of such shares would or might involve the contravention of the laws of any territory or that for any other reason the offer should not be made to them or in respect of such shares; |
| (h) | unless the Board decides otherwise or the rules of a relevant system require otherwise, any new shares which a holder has elected to receive instead of cash in respect of some or all of his or her dividend will be: |
| (i) | shares in uncertificated form if the corresponding elected shares were uncertificated shares on the record date for that dividend; and |
| (ii) | shares in certificated form if the corresponding elected shares were shares in certificated form on the record date for that dividend; |
| (i) | the Board may establish or vary a procedure for election mandates in respect of future rights of election and may determine that every duly effected election in respect of any shares shall be binding on every successor in title to the holder; |
| (j) | the dividend (or that part of the dividend in respect of which a right of election has been offered) shall not be payable on shares in respect of which an election has been duly made (elected shares) and instead additional shares shall be allotted to the holders of the elected shares on the basis of allotment determined as stated above. For such purpose the Board may capitalise, out of any amount for the time being standing to the credit of any reserve or fund (including any share premium account or capital redemption reserve) or of any of the profits which could otherwise have been applied in paying dividends in cash as the Board may determine, a sum equal to the aggregate nominal amount of the additional shares to be allotted on such basis and apply it in paying up in full the appropriate number of unissued shares for allotment and distribution to the holders of the elected shares on such basis. The Board may do all acts and things considered necessary or expedient to give effect to any such capitalisation; |
| (k) | the Board may decide how any costs relating to the new shares available in place of a cash dividend will be met, including to deduct an amount from the entitlement of a holder of shares under this Article; |
| (l) | the additional shares so allotted shall rank pari passu in all respects with each other and with the fully paid shares in issue on the record date for the dividend in respect of which the right of election has been offered, except that they will not rank for any dividend or other distribution or other entitlement which has been declared, paid or made by reference to such record date; |
| (m) | the Board may terminate, suspend, or amend any offer of the right to elect to receive shares in lieu of any cash dividend at any time and generally may implement any scrip dividend scheme on such terms and conditions as the Board may determine and take such other action as the Board may deem necessary or desirable in respect of any such scheme; and |
| (n) | the Board may do all acts and things which they consider necessary or expedient to give effect to any such capitalisation, and may authorise any person to enter on behalf of all the members interested into an agreement with the Company providing for such capitalisation and incidental matters and any agreement so made shall be binding on all concerned. |
| 133. | Capitalisation of reserves |
| 133.1 | The Board may, with the authority of an ordinary resolution of the Company: |
| (a) | subject as provided in this Article, resolve to capitalise any undivided profits of the Company not required for paying any preferential dividend (whether or not they are available for distribution) or any sum standing to the credit of any reserve or fund of the Company which is available for distribution or standing to the credit of the share premium account or capital redemption reserve or other undistributable reserve; |
| (b) | appropriate the sum resolved to be capitalised to the members in proportion to the nominal amounts of the shares (whether or not fully paid) held by them respectively which would entitle them to participate in a distribution of that sum if the shares were fully paid and the sum were then distributable and were distributed by way of dividend and apply such sum on their behalf either in or towards paying up the amounts, if any, for the time being unpaid on any shares held by them respectively, or in paying up in full unissued shares or debentures of the Company of a nominal amount equal to that sum, and allot the shares or debentures credited as fully paid to those members or as they may direct, in those proportions, or partly in one way and partly in the other, provided that: |
| (i) | the share premium account, the capital redemption reserve, any other undistributable reserve and any profits which are not available for distribution may, for the purposes of this Article, only be applied in paying up in full shares to be allotted to members credited as fully paid; |
| (ii) | the Company will also be entitled to participate in the relevant distribution in relation to any shares of the relevant class held by it as treasury shares and the proportionate entitlement of the relevant class of members to the distribution will be calculated accordingly; and |
| (iii) | in a case where any sum is applied in paying amounts for the time being unpaid on any shares of the Company or in paying up in full debentures of the Company, the amount of the net assets of the Company at that time is not less than the aggregate of the called up share capital of the Company and its undistributable reserves as shown in the latest audited accounts of the Company or such other accounts as may be relevant and would not be reduced below that aggregate by the payment of it; |
| (c) | resolve that any shares so allotted to any member in respect of a holding by him or her of any partly paid shares shall, so long as such shares remain partly paid, rank for dividends only to the extent that such partly paid shares rank for dividends; |
| (d) | make such provision by the issue of fractional certificates (or by ignoring fractions or by accruing the benefit of it to the Company rather than to the members concerned) or by payment in cash or otherwise as it thinks fit in the case of shares or debentures becoming distributable in fractions; |
| (e) | authorise any person to enter on behalf of such members concerned into an agreement with the Company providing for either: |
| (i) | the allotment to them respectively, credited as fully paid up, of any shares or debentures to which they may be entitled on such capitalisation; or |
| (ii) | the payment up by the Company on behalf of such members by the application of their respective proportions of the reserves or profits resolved to be capitalised, of the amounts or any part of the amounts remaining unpaid on their existing shares, |
(any agreement made under such authority being effective and binding on all such members); and
| (f) | generally do all acts and things required to give effect to such resolution. |
| 133.2 | Where, pursuant to an employees’ share scheme (within the meaning of section 1166 of the Act) or any similar scheme under which participation is extended to non-executive Directors or consultants providing services to the Company or any of its subsidiaries: |
| (a) | the Company has granted options to subscribe for shares on terms which provide (inter alia) for adjustments to the subscription price payable on the exercise of such options or to the number of shares to be allotted upon such exercise in the event of any increase or reduction in or other reorganisation of the Company’s issued share capital and an otherwise appropriate adjustment would result in the subscription price for any share being less than its nominal value, then the Board may, on the exercise of any of the options concerned and payment of the subscription price which would have applied had such adjustment been made, capitalise any such profits or other sum as is mentioned in Article 133.1(a) to the extent necessary to pay up the unpaid balance of the nominal value of the shares which fall to be allotted on the exercise of such options and apply such amount in paying up such balance and allot shares fully paid accordingly; |
| (b) | the Company has granted (or assumed liability to satisfy) rights to subscribe for shares (whether in the form of stock options, stock units, restricted stock, stock appreciation rights, performance shares and units, dividend equivalent rights or otherwise) then the Board may, in connection with the issue of shares, capitalise any such profits or other sum as is mentioned in Article 133.1 to the extent necessary to pay up the unpaid balance of the nominal value of the shares which fall to be issued in connection with such rights to subscribe and apply such amount in paying up such balance and allot shares fully paid accordingly; and |
| (c) | the provisions of Article 133.1(a) to (f) shall apply with the necessary alterations to this Article 133.2. |
| 134. | Record dates |
| 134.1 | Notwithstanding any other provision of these Articles but without prejudice to the rights attached to any shares and subject always to the Act, the Company or the Board may by resolution specify any date (record date) as the date at the close of business (or such other time as the Board may determine) on which persons registered as the holders of shares or other securities shall be entitled to receipt of any dividend, distribution, interest, allotment, issue, notice, information, document or circular. Such record date may be before, on or after the date on which the dividend, distribution, interest, allotment, issue, notice, information, document or circular is declared, made, paid, given, or served. |
| 134.2 | In the absence of a record date being fixed, entitlement to any dividend, distribution, interest, allotment, issue, notice, information, document or circular shall be determined by reference to the date on which the dividend is declared, the distribution allotment or issue is made or the notice, information, document or circular made, given or served. |
| 135. | Inspection of records |
No member (other than a Director) shall have any right to inspect any accounting record or other document of the Company unless he or she is authorised to do so by law, by order of a court of competent jurisdiction, by the Board or by ordinary resolution of the Company.
| 136. | Account to be sent to members |
| 136.1 | In respect of each financial year, a copy of the Company’s annual accounts, the strategic report, the Directors’ report, the Directors’ remuneration report, the auditor’s report on those accounts and on the auditable part of the Directors’ remuneration report shall be sent or supplied to: |
| (a) | every member (whether or not entitled to receive notices of general meetings); |
| (b) | every holder of debentures (whether or not entitled to receive notice of general meetings); and |
| (c) | every other person who is entitle to receive notice of general meetings, |
not less than twenty-one clear days before the date of the meeting at which copies of those documents are to be laid in accordance with the Act.
| 136.2 | This Article does not require copies of the documents to which it applies to be sent or supplied to: |
| (a) | a member or holder of debentures of whose address the Company is unaware; or |
| (b) | more than one of the joint holders of shares or debentures. |
| 136.3 | The Board may determine that persons entitled to receive a copy of the Company’s annual accounts, the strategic report, the Directors’ report, the Directors’ remuneration report, the auditor’s report on those accounts and on the auditable part of the Directors’ remuneration report are those persons entered on the Register at the close of business on a day determined by the Board, provided that the day determined by the Board may not be more than twenty-one days before the day that the relevant copies are being sent. |
| 136.4 | Where permitted by the Act, a strategic report with supplementary material in the form and containing the information prescribed by the Act may be sent or supplied to a person so electing in place of the documents required to be sent or supplied by Article 136.1. |
| 137. | Service of Notices |
| 137.1 | The Company can send, deliver or serve any notice or other document, including a share certificate, to or on a member: |
| (a) | personally; |
| (b) | by sending it through the postal system addressed to the member at his, her or its registered address or by leaving it at that address addressed to the member; |
| (c) | through a relevant system, where the notice or document relates to uncertificated shares; |
| (d) | where appropriate, by sending or supplying it in electronic form to an address notified by the member to the Company for that purpose; |
| (e) | where appropriate, by making it available on a website and notifying the member of its availability in accordance with this Article; or |
| (f) | by any other means authorised in writing by the member. |
| 137.2 | In the case of joint holders of a share: |
| (a) | service, sending or supply of any notice, document or other information on or to one of the joint holders shall for all purposes be deemed a sufficient service on, sending or supplying to all the joint holders; and |
| (b) | anything to be agreed or specified in relation to any notice, document or other information to be served on, sent or supplied to them may be agreed or specified by any one of the joint holders and the agreement or specification of the first named in the Register shall be accepted to the exclusion of that of the other joint holders. |
| 137.3 | Where a member (or, in the case of a joint holders, the person first named in the Register) has a registered address outside the United Kingdom but has (i) notified the Company of an address within the United Kingdom at which notices, documents or other information may be given to him, her or it; or (ii) given to the Company an address for the purposes of communications by electronic means at which notices, documents or other information may be served, sent or supplied to him, her or it, he, she or it shall be entitled to have notices served, sent or supplied to him or her at such address or, where applicable, the Company may make them available on a website and notify the holder of that address. Otherwise no such member shall be entitled to receive any notice, document or other information from the Company. |
| 137.4 | If on three consecutive occasions any notice, document or other information has been sent to any member at his, her or its registered address or his, her or its address for the service of notices (by electronic means or otherwise) but has been returned undelivered, such member shall not be entitled to receive notices, documents or other information from the Company until he or she shall have communicated with the Company and supplied in writing a new registered address or address within the United Kingdom for the service of notices or has informed the Company of an address for the service of notices and the sending or supply of documents and other information in electronic form. For these purposes, any notice, document or other information served, sent or supplied by post shall be treated as returned undelivered if the notice, document or other information is served, sent or supplied back to the Company (or its agents) and a notice, document or other information served, sent or supplied in electronic form shall be treated as returned undelivered if the Company (or its agents) receives notification that the notice, document or other information was not delivered to the address to which it was served, sent or supplied. |
| 137.5 | The Company may at any time and in its sole discretion choose to serve, send or supply notices, documents or other information in hard copy form alone to some or all of the members. |
| 138. | Hard copy form |
Any document, information or notice is validly sent or supplied by the Company in hard copy form if it is handed to the intended recipient or sent or supplied by hand or through the post in a prepaid envelope:
| (a) | to an address specified for the purpose by the intended recipient; |
| (b) | if the intended recipient is a company, to its registered office; |
| (c) | to the address shown in the Company’s Register; |
| (d) | to any address to which any provision of the Companies Acts authorises it to be sent or supplied; or |
| (e) | if the Company is unable to obtain an address falling within paragraphs (a) to (d), to the last address known to the Company of the intended recipient. |
| 139. | Electronic form |
Any document, information or notice is validly sent or supplied by the Company in electronic form:
| (a) | to a person if that person has agreed (generally or specifically) that the document, information or notice may be sent or supplied in that form and has not revoked that agreement; or |
| (b) | to a company that is deemed to have so agreed by the Companies Acts. |
| 140. | Electronic means |
Any document, information or notice is validly sent or supplied by the Company by electronic means if it is sent or supplied:
| (a) | to an address specified for the purpose by the intended recipient (generally or specifically); or |
| (b) | where the intended recipient is a company, to an address deemed by the Companies Acts to have been so specified. |
| 141. | Website |
Any document, information or notice is validly sent or supplied by the Company to a person by being made available on a website if:
| (a) | the person has agreed (generally or specifically) that the document, information or notice may be sent or supplied to him or her in that manner, or he or she is taken to have so agreed under Schedule 5 of the Act, and in either case he or she has not revoked that agreement; |
| (b) | the Company has notified the intended recipient of: |
| (i) | the presence of the document, information or notice on the website; |
| (ii) | the address of the website; |
| (iii) | the place on the website where it may be accessed; |
| (iv) | how to access the document, information or notice; and |
| (v) | any other information prescribed by the Companies Acts or any other provisions of law including, when the document, information or notice is a notice of meeting, that fact, the place, date and time of the meeting and whether the meeting is an annual general meeting; and |
| (c) | the document, information or notice is available on the website throughout the period specified by any applicable provision of the Companies Acts or, if no such period is specified, the period of twenty-eight days starting on the date on which the notification referred to in paragraph (b) above is sent to the relevant person. |
| 142. | Sending or supplying any document, information or notice by any other means |
Any document, information or notice that is sent or supplied otherwise than in hard copy form or electronic form or by means of a website is validly sent or supplied if it is sent or supplied in a form or manner that has been agreed by the intended recipient.
| 143. | Presence at meeting evidence in itself of receipt of notice |
A member present either in person or by proxy, or in the case of a corporate member by a duly authorised representative, at any meeting of the Company or of the holders of any class of shares shall be deemed to have received notice of the meeting and, where required, of the purposes for which it was called.
| 144. | Notice on person entitled by transmission |
The Company may give notice to the person entitled to a share because of the death or bankruptcy of a member or otherwise by operation of law, by sending or delivering it in any manner authorised by these Articles for the giving of notice to a member, addressed to that person by name, or by the title of representative of the deceased or trustee of the bankrupt or representative by operation of law or by any like description, at the address (if any) within the United Kingdom supplied for the purpose by the person claimed to be so entitled or to which notices may be sent in electronic form. Until such an address has been so supplied, a notice may be given in any manner in which it might have been given if the death or bankruptcy or operation of law had not occurred. This shall apply whether or not the Company has notice of the death or bankruptcy or other event.
| 145. | Record date for service |
Any notice, document or other information may be served, sent or supplied by the Company by reference to the register as it stands at any time not more than fifteen days before the date of service, sending or supplying. No change in the register after that time shall invalidate that service, sending or supply. Where any notice, document or other information is served on, sent or supplied to any person in respect of a share in accordance with these Articles, no person deriving any title or interest in that share shall be entitled to any further service, sending or supplying of that notice, document or other information.
| 146. | Evidence of service |
| 146.1 | Any notice, document or other information, addressed to a member at his, her or its registered address or address for service in the United Kingdom shall, if served, sent or supplied by first class post, be deemed to have been served or delivered on the day after the day when it was put in the post (or, where second class post is employed, on the second day after the day when it was put in the post). Proof that an envelope containing the notice, document or other information was properly addressed and put into the post as a prepaid letter shall be conclusive evidence that the notice was given. |
| 146.2 | Any notice, document or other information not served, sent or supplied by post but delivered or left at a registered address or address for service in the United Kingdom (other than an address for the purposes of communications by electronic means) shall be deemed to have been served or delivered on the day on which it was so delivered or left. |
| 146.3 | Any notice, document or other information, if served, sent or supplied by electronic means shall be deemed to have been received on the day on which the electronic communication was sent by or on behalf of the Company notwithstanding that the Company subsequently sends such notice, document or other information in hard copy form by post. Any notice, document or other information made available on a website shall be deemed to have been received on the day on which the notice, document or other information was first made available on the website or, if later, when a notice of availability is received or deemed to have been received pursuant to this Article. Proof that the notice, document or other information was properly addressed shall be conclusive evidence that the notice by electronic means was given. |
| 146.4 | Any notice, document or other information served, sent or supplied by the Company by means of a relevant system shall be deemed to have been received when the Company or any sponsoring system-participant acting on its behalf sends the issuer-instruction relating to the notice, document or other information. |
| 146.5 | Any notice, document or other information served, sent or supplied by the Company by any other means authorised in writing by the member concerned shall be deemed to have been received when the Company has carried out the action it has been authorised to take for that purpose. |
| 147. | Notice when post not available |
If at any time by reason of the suspension, interruption or curtailment of postal services within the United Kingdom the Company is unable effectively to convene a general meeting by notices sent through the post, the Company need only give notice of a general meeting to those members with whom the Company can communicate by electronic means and who have provided the Company with an address for this purpose. The Company shall also advertise the notice in at least one national newspaper published in the United Kingdom and make it available on its website from the date of such advertisement until the conclusion of the meeting or any adjournment of it. In any such case the Company shall send confirmatory copies of the notice by post to those members to whom notice cannot be given by electronic means if, at least seven days prior to the meeting, the posting of notices to addresses throughout the United Kingdom again becomes practicable.
| 148. | Validation of documents in electronic form |
| 148.1 | Where a document is required under these Articles to be signed by a member or any other person, if the document is in electronic form, then in order to be valid the document must: |
| (a) | incorporate the electronic signature, or personal identification details (which may be details previously allocated by the Company), of that member or other person, in such form as the Board may approve; or |
| (b) | be accompanied by such other evidence as the Board may require in order to be satisfied that the document is genuine. |
| 148.2 | The Company may designate mechanisms for validating any such document and a document not validated by the use of any such mechanisms shall be deemed as having not been received by the Company In the case of any document or information relating to a meeting, an instrument of proxy or invitation to appoint a proxy, any validation requirements shall be specified in the relevant notice of meeting in accordance with Articles 49 and 70. |
| 149. | Winding up |
If the Company is wound up and subject to the rights and restrictions attached to any share or classes of shares, the liquidator may, with the sanction of a special resolution and any other sanction required by law, divide among the members in specie the whole or any part of the assets of the Company and may, for that purpose, value any assets and determine how the division shall be carried out as between the members or different classes of members. The liquidator may, with the like sanction(s), vest the whole or any part of the assets in trustees upon such trusts for the benefit of the members as he, she or it may with the like sanction determine. Where the liquidator divides or transfers any assets in pursuance of the powers in this Article 149, no member shall be compelled to accept any assets upon which there is a liability.
| 150. | Indemnity and insurance |
| 150.1 | In this Article: |
| (a) | companies are associated if one is a subsidiary of the other or both are subsidiaries of the same body corporate; |
| (b) | a relevant officer means any Director or other officer or former Director or other officer of the Company or an associated company (including any company which is a trustee of an occupational pension scheme (as defined by section 235(6) of the Act), but excluding in each case any person engaged by the Company (or associated company) as auditor (whether or not he or she is also a Director or other officer), to the extent he or she acts in his or her capacity as auditor); and |
| (c) | relevant loss means any loss or liability which has been or may be incurred by a relevant officer in connection with that relevant officer’s duties or powers in relation to the company, any associated company or any pension fund or employees’ share scheme of the company or associated company. |
| 150.2 | Subject to Article 150.3, but without prejudice to any indemnity to which a relevant officer is otherwise entitled: |
| (a) | each relevant officer shall be indemnified out of the Company’s assets against all relevant loss and in relation to the Company’s (or any associated company’s) activities as trustee of an occupational pension scheme (as defined in section 235(6) of the Act), including any liability incurred by him or her in defending any civil or criminal proceedings, in which judgment is given in his or her favour or in which he or she is acquitted or the proceedings are otherwise disposed of without any finding or admission of any material breach of duty on his or her part or in connection with any application in which the court grants him or her, in his or her capacity as a relevant officer, relief from liability for negligence, default, breach of duty or breach of trust in relation to the Company’s (or any associated company’s) affairs; and |
| (b) | the Company may provide any relevant officer with funds to meet expenditure incurred or to be incurred by him or her in connection with any proceedings or application referred to in Article 150.2(a) and otherwise may take any action to enable any such relevant officer to avoid incurring such expenditure. |
| 150.3 | This Article does not authorise any indemnity which would be prohibited or rendered void by any provision of the Companies Acts or by any other provision of law. |
| 150.4 | The Directors may decide to purchase and maintain insurance, at the expense of the Company, for the benefit of any relevant officer in respect of any relevant loss. |
| 150.5 | Where a relevant officer is indemnified against a liability in accordance with this Article, the indemnity extends to each cost, charge, loss, expense and liability incurred by him or her in relation to that liability. |
| 151. | Exclusive jurisdiction |
| 151.1 | Save in respect of any cause of action arising under the Securities Act or the Exchange Act, unless the Company by ordinary resolution consents to the selection of an alternative forum, the courts of England and Wales shall be the exclusive forum for the resolution of: |
| (a) | any derivative action or proceeding brought on behalf of the Company; |
| (b) | any action or proceeding asserting a claim of breach of fiduciary duty owed by any director, officer or other employee to the Company; |
| (c) | any action or proceeding asserting a claim arising out of any provision of the Companies Acts or these Articles; or |
| (d) | any action or proceeding asserting a claim or otherwise related to the affairs of the Company. |
| 151.2 | Unless the Company by ordinary resolution consents to the selection of an alternative forum in the United States, the United States District Court for the Southern District of New York shall be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act or the Exchange Act. |
| 151.3 | Any person or entity purchasing or otherwise acquiring any interest in the Company’s shares shall be deemed to have notice of and to have consented to the provisions of this Article 151. |
Exhibit 2.2
DEPOSIT AGREEMENT
by and among
Immunocore Holdings PLC
and
CITIBANK, N.A.,
as Depositary,
and
THE HOLDERS AND BENEFICIAL OWNERS OF
AMERICAN DEPOSITARY SHARES
ISSUED HEREUNDER
Dated as of February 9, 2021
TABLE OF CONTENTS
| ARTICLE I | ||
| DEFINITIONS | 1 | |
| Section 1.1 | “ADS Record Date” | 1 |
| Section 1.2 | “Affiliate” | 1 |
| Section 1.3 | “American Depositary Receipt(s)”, “ADR(s)” and “Receipt(s)” | 1 |
| Section 1.4 | “American Depositary Share(s)” and “ADS(s)” | 2 |
| Section 1.5 | “Beneficial Owner” | 2 |
| Section 1.6 | “Certificated ADS(s)” | 3 |
| Section 1.7 | “Citibank” | 3 |
| Section 1.8 | “Commission” | 3 |
| Section 1.9 | “Company” | 3 |
| Section 1.10 | “CREST” | 3 |
| Section 1.11 | “Custodian” | 3 |
| Section 1.12 | “Deliver” and “Delivery” | 3 |
| Section 1.13 | “Deposit Agreement” | 3 |
| Section 1.14 | “Depositary” | 4 |
| Section 1.15 | “Deposited Property” | 4 |
| Section 1.16 | “Deposited Securities” | 4 |
| Section 1.17 | “Dollars” and “$” | 4 |
| Section 1.18 | “DTC” | 4 |
| Section 1.19 | “DTC Participant” | 4 |
| Section 1.20 | “Exchange Act” | 4 |
| Section 1.21 | “Foreign Currency” | 4 |
| Section 1.22 | “Full Entitlement ADR(s)”, “Full Entitlement ADS(s)” and “Full Entitlement Share(s)” | 4 |
| Section 1.23 | “Holder(s)” | 5 |
| Section 1.24 | “Partial Entitlement ADR(s)”, “Partial Entitlement ADS(s)” and “Partial Entitlement Share(s)” | 5 |
| Section 1.25 | “Pounds”, “Pence” and “£” | 5 |
| Section 1.26 | “Principal Office” | 5 |
| Section 1.27 | “Registrar” | 5 |
| Section 1.28 | “Restricted Securities” | 5 |
| Section 1.29 | “Restricted ADR(s)”, “Restricted ADS(s)” and “Restricted Shares” | 5 |
| Section 1.30 | “Securities Act” | 6 |
| Section 1.31 | “Share Registrar” | 6 |
| Section 1.32 | “Shares” | 6 |
| Section 1.33 | “Uncertificated ADS(s)” | 6 |
| Section 1.34 | “United States” and “U.S.” | 6 |
| ARTICLE II | ||
| APPOINTMENT OF DEPOSITARY; FORM OF RECEIPTS; | ||
| DEPOSIT OF SHARES; EXECUTION AND | ||
| DELIVERY, TRANSFER AND SURRENDER OF RECEIPTS | 6 | |
| Section 2.1 | Appointment of Depositary | 6 |
| Section 2.2 | Form and Transferability of ADSs | 6 |
| Section 2.3 | Deposit of Shares | 8 |
| Section 2.4 | Registration and Safekeeping of Deposited Securities | 10 |
| Section 2.5 | Issuance of ADSs | 10 |
| Section 2.6 | Transfer, Combination and Split-up of ADRs | 11 |
| Section 2.7 | Surrender of ADSs and Withdrawal of Deposited Securities | 11 |
| Section 2.8 | Limitations on Execution and Delivery, Transfer, etc. of ADSs; Suspension of Delivery, Transfer, etc | 13 |
| Section 2.9 | Lost ADRs, etc | 13 |
| Section 2.10 | Cancellation and Destruction of Surrendered ADRs; Maintenance of Records | 14 |
| Section 2.11 | Escheatment | 14 |
| Section 2.12 | Partial Entitlement ADSs | 14 |
| Section 2.13 | Certificated/Uncertificated ADSs | 15 |
| Section 2.14 | Restricted ADSs | 16 |
| ARTICLE III | ||
| CERTAIN OBLIGATIONS OF HOLDERS | ||
| AND BENEFICIAL OWNERS OF ADSs | 18 | |
| Section 3.1 | Proofs, Certificates and Other Information | 18 |
| Section 3.2 | Liability for Taxes and Other Charges | 18 |
| Section 3.3 | Representations and Warranties on Deposit of Shares | 19 |
| Section 3.4 | Compliance with Information Requests | 19 |
| Section 3.5 | Ownership Restrictions | 20 |
| Section 3.6 | Reporting Obligations and Regulatory Approvals | 20 |
| ARTICLE IV | ||
| THE DEPOSITED SECURITIES | 21 | |
| Section 4.1 | Cash Distributions | 21 |
| Section 4.2 | Distribution in Shares | 22 |
| Section 4.3 | Elective Distributions in Cash or Shares | 23 |
| Section 4.4 | Distribution of Rights to Purchase Additional ADSs | 24 |
| Section 4.5 | Distributions Other Than Cash, Shares or Rights to Purchase Shares | 25 |
| Section 4.6 | Distributions with Respect to Deposited Securities in Bearer Form | 26 |
| Section 4.7 | Redemption | 26 |
| Section 4.8 | Conversion of Foreign Currency | 27 |
| Section 4.9 | Fixing of ADS Record Date | 28 |
| Section 4.10 | Voting of Deposited Securities | 28 |
| Section 4.11 | Changes Affecting Deposited Securities | 30 |
| Section 4.12 | Available Information | 31 |
| Section 4.13 | Reports | 31 |
| Section 4.14 | List of Holders | 31 |
| Section 4.15 | Taxation | 31 |
| ARTICLE V | ||
| THE DEPOSITARY, THE CUSTODIAN AND THE COMPANY | 33 | |
| Section 5.1 | Maintenance of Office and Transfer Books by the Registrar | 33 |
| Section 5.2 | Exoneration | 33 |
| Section 5.3 | Standard of Care | 34 |
| Section 5.4 | Resignation and Removal of the Depositary; Appointment of Successor Depositary | 35 |
| Section 5.5 | The Custodian | 36 |
| Section 5.6 | Notices and Reports | 36 |
| Section 5.7 | Issuance of Additional Shares, ADSs etc | 37 |
| Section 5.8 | Indemnification | 38 |
| Section 5.9 | ADS Fees and Charges | 39 |
| Section 5.10 | Restricted Securities Owners | 40 |
| ARTICLE VI | ||
| AMENDMENT AND TERMINATION | 40 | |
| Section 6.1 | Amendment/Supplement | 40 |
| Section 6.2 | Termination | 41 |
| ARTICLE VII | ||
| MISCELLANEOUS | 43 | |
| Section 7.1 | Counterparts | 43 |
| Section 7.2 | No Third Party Beneficiaries/Acknowledgments | 43 |
| Section 7.3 | Severability | 43 |
| Section 7.4 | Holders and Beneficial Owners as Parties; Binding Effect | 44 |
| Section 7.5 | Notices | 44 |
| Section 7.6 | Governing Law and Jurisdiction | 45 |
| Section 7.7 | Assignment | 46 |
| Section 7.8 | Compliance with, and No Disclaimer under, U.S. Securities Laws | 46 |
| Section 7.9 | English Law References | 47 |
| Section 7.10 | Titles and References | 47 |
| EXHIBITS | ||
| Form of ADR. | A-1 | |
| Fee Schedule. | B-1 | |
DEPOSIT AGREEMENT
DEPOSIT AGREEMENT, dated as of February 9, 2021, by and among (i) Immunocore Holdings plc, a public limited company incorporated under the laws of England and Wales, and its successors (the “Company”), (ii) CITIBANK, N.A., a national banking association organized under the laws of the United States of America (“Citibank”) acting in its capacity as depositary, and any successor depositary hereunder (Citibank in such capacity, the “Depositary”), and (iii) all Holders and Beneficial Owners of American Depositary Shares issued hereunder (all such capitalized terms as hereinafter defined).
W I T N E S S E T H T H A T:
WHEREAS, the Company desires to establish with the Depositary an ADR facility to provide for the deposit of the Shares (as hereinafter defined) and the creation of American Depositary Shares representing the Shares so deposited and for the execution and Delivery (as hereinafter defined) of American Depositary Receipts (as hereinafter defined) evidencing such American Depositary Shares; and
WHEREAS, the Depositary is willing to act as the Depositary for such ADR facility upon the terms set forth in the Deposit Agreement (as hereinafter defined); and
WHEREAS, any American Depositary Receipts issued pursuant to the terms of the Deposit Agreement are to be substantially in the form of Exhibit A attached hereto, with appropriate insertions, modifications and omissions, as hereinafter provided in the Deposit Agreement.
NOW, THEREFORE, for good and valuable consideration, the receipt and sufficiency of which are hereby acknowledged, the parties hereto agree as follows:
ARTICLE I
DEFINITIONS
All capitalized terms used, but not otherwise defined, herein shall have the meanings set forth below, unless otherwise clearly indicated:
Section 1.1 “ADS Record Date” shall have the meaning given to such term in Section 4.9.
Section 1.2 “Affiliate” shall have the meaning assigned to such term by the Commission (as hereinafter defined) under Regulation C promulgated under the Securities Act (as hereinafter defined), or under any successor regulation thereto.
Section 1.3 “American Depositary Receipt(s)”, “ADR(s)” and “Receipt(s)” shall mean the certificate(s) issued by the Depositary to evidence the American Depositary Shares issued under the terms of the Deposit Agreement in the form of Certificated ADS(s) (as hereinafter defined), as such ADRs may be amended from time to time in accordance with the provisions of the Deposit Agreement. An ADR may evidence any number of ADSs and may, in the case of ADSs held through a central depository such as DTC, be in the form of a “Balance Certificate.”
Section 1.4 “American Depositary Share(s)” and “ADS(s)” shall mean the rights and interests in the Deposited Property (as hereinafter defined) granted to the Holders and Beneficial Owners pursuant to the terms and conditions of the Deposit Agreement and, if issued as Certificated ADS(s) (as hereinafter defined), the ADR(s) issued to evidence such ADSs. ADS(s) may be issued under the terms of the Deposit Agreement in the form of (a) Certificated ADS(s) (as hereinafter defined), in which case the ADS(s) are evidenced by ADR(s), or (b) Uncertificated ADS(s) (as hereinafter defined), in which case the ADS(s) are not evidenced by ADR(s) but are reflected on the direct registration system maintained by the Depositary for such purposes under the terms of Section 2.13. Unless otherwise specified in the Deposit Agreement or in any ADR, or unless the context otherwise requires, any reference to ADS(s) shall include Certificated ADS(s) and Uncertificated ADS(s), individually or collectively, as the context may require. Each ADS shall represent the right to receive, and to exercise the beneficial ownership interests in, the number of Shares specified in the form of ADR attached hereto as Exhibit A (as amended from time to time) that are on deposit with the Depositary and/or the Custodian, subject, in each case, to the terms and conditions of the Deposit Agreement and the applicable ADR (if issued as a Certificated ADS), until there shall occur a distribution upon Deposited Securities referred to in Section 4.2 or a change in Deposited Securities referred to in Section 4.11 with respect to which additional ADSs are not issued, and thereafter each ADS shall represent the right to receive, and to exercise the beneficial ownership interests in, the applicable Deposited Property on deposit with the Depositary and the Custodian determined in accordance with the terms of such Sections, subject, in each case, to the terms and conditions of the Deposit Agreement and the applicable ADR (if issued as a Certificated ADS). In addition, the ADS(s)-to-Share(s) ratio is subject to amendment as provided in Articles IV and VI of the Deposit Agreement (which may give rise to Depositary fees).
Section 1.5 “Beneficial Owner” shall mean, as to any ADS, any person or entity having a beneficial interest deriving from the ownership of such ADS. Notwithstanding anything else contained in the Deposit Agreement, any ADR(s) or any other instruments or agreements relating to the ADSs and the corresponding Deposited Property, the Depositary, the Custodian and their respective nominees are intended to be, and shall at all times during the term of the Deposit Agreement be, the record holders only of the Deposited Property represented by the ADSs for the benefit of the Holders and Beneficial Owners of the corresponding ADSs. The Depositary, on its own behalf and on behalf of the Custodian and their respective nominees, disclaims any beneficial ownership interest in the Deposited Property held on behalf of the Holders and Beneficial Owners of ADSs. The beneficial ownership interests in the Deposited Property are intended to be, and shall at all times during the term of the Deposit Agreement continue to be, vested in the Beneficial Owners of the ADSs representing the Deposited Property. The beneficial ownership interests in the Deposited Property shall, unless otherwise agreed by the Depositary, be exercisable by the Beneficial Owners of the ADSs only through the Holders of such ADSs, by the Holders of the ADSs (on behalf of the applicable Beneficial Owners) only through the Depositary, and by the Depositary (on behalf of the Holders and Beneficial Owners of the corresponding ADSs) directly, or indirectly through the Custodian or their respective nominees, in each case upon the terms of the Deposit Agreement and, if applicable, the terms of the ADR(s) evidencing the ADSs. A Beneficial Owner of ADSs may or may not be the Holder of such ADSs. A Beneficial Owner shall be able to exercise any right or receive any benefit hereunder solely through the person who is the Holder of the ADSs owned by such Beneficial Owner. Unless otherwise identified to the Depositary, a Holder shall be deemed to be the Beneficial Owner of all the ADSs registered in his/her/its name. The manner in which a Beneficial Owner holds ADSs (e.g., in a brokerage account vs. as registered holder) may affect the rights and obligations of, the manner in which, and the extent to which, services are made available to, Beneficial Owners pursuant to the terms of the Deposit Agreement.
Section 1.6 “Certificated ADS(s)”shall have the meaning set forth in Section 2.13.
Section 1.7 “Citibank” shall mean Citibank, N.A., a national banking association organized under the laws of the United States of America, and its successors.
Section 1.8 “Commission” shall mean the Securities and Exchange Commission of the United States or any successor governmental agency thereto in the United States.
Section 1.9 “Company” shall mean Immunocore Holdings plc, a public limited company incorporated under the laws of England and Wales, and its successors.
Section 1.10 “CREST” shall mean the system for the paperless settlement of trades in securities and the holding of uncertificated securities operated by Euroclear UK & Ireland Limited in accordance with the Uncertificated Securities Regulations 2001 (SI 2001 No. 3755), as amended from time to time, or any successor thereto.
Section 1.11 “Custodian” shall mean (i) as of the date hereof, Citibank, N.A. (London), having its principal office at 25 Canada Square, Canary Wharf, London E14 5LB, United Kingdom, as the custodian of Deposited Property for the purposes of the Deposit Agreement, (ii) Citibank, N.A., acting as custodian of Deposited Property pursuant to the Deposit Agreement, and (iii) any other entity that may be appointed by the Depositary pursuant to the terms of Section 5.5 as successor, substitute or additional custodian hereunder. The term “Custodian” shall mean any Custodian individually or all Custodians collectively, as the context requires.
Section 1.12 “Deliver” and “Delivery” shall mean (x) when used in respect of Shares and other Deposited Securities, either (i) the physical delivery of the certificate(s) representing such securities, or (ii) the book-entry transfer and recordation of such securities on the books of the Share Registrar (as hereinafter defined) or in the book-entry settlement of CREST, and (y) when used in respect of ADSs, either (i) the physical delivery of ADR(s) evidencing the ADSs, or (ii) the book-entry transfer and recordation of ADSs on the books of the Depositary or any book-entry settlement system in which the ADSs are settlement-eligible.
Section 1.13 “Deposit Agreement” shall mean this Deposit Agreement and all exhibits hereto, as the same may from time to time be amended and supplemented from time to time in accordance with the terms of the Deposit Agreement.
Section 1.14 “Depositary” shall mean Citibank, N.A., a national banking association organized under the laws of the United States, in its capacity as depositary under the terms of the Deposit Agreement, and any successor depositary hereunder.
Section 1.15 “Deposited Property” shall mean the Deposited Securities and any cash and other property held on deposit by the Depositary and the Custodian in respect of the ADSs or the Deposited Securities under the terms of the Deposit Agreement, subject, in the case of cash, to the provisions of Section 4.8. All Deposited Property shall be held by the Custodian, the Depositary and their respective nominees for the benefit of the Holders and Beneficial Owners of the ADSs representing the Deposited Property. The Deposited Property is not intended to, and shall not, constitute proprietary assets of the Depositary, the Custodian or their nominees. Beneficial ownership in the Deposited Property is intended to be, and shall at all times during the term of the Deposit Agreement continue to be, vested in the Beneficial Owners of the ADSs representing the Deposited Property.
Section 1.16 “Deposited Securities” shall mean the Shares and any other securities held on deposit by the Custodian from time to time in respect of the ADSs under the Deposit Agreement and constituting Deposited Property.
Section 1.17 “Dollars” and “$” shall refer to the lawful currency of the United States.
Section 1.18 “DTC” shall mean The Depository Trust Company, a national clearinghouse and the central book-entry settlement system for securities traded in the United States and, as such, the custodian for the securities of DTC Participants (as hereinafter defined) maintained in DTC, and any successor thereto.
Section 1.19 “DTC Participant” shall mean any financial institution (or any nominee of such institution) having one or more participant accounts with DTC for receiving, holding and delivering the securities and cash held in DTC. A DTC Participant may or may not be a Beneficial Owner. If a DTC Participant is not the Beneficial Owner of the ADSs credited to its account at DTC, or of the ADSs in respect of which the DTC Participant is otherwise acting, such DTC Participant shall be deemed, for all purposes hereunder, to have all requisite authority to act on behalf of the Beneficial Owner(s) of the ADSs credited to its account at DTC or in respect of which the DTC Participant is so acting. A DTC Participant, upon acceptance in any one of its DTC accounts of any ADSs (or any interest therein) issued in accordance with the terms and conditions of the Deposit Agreement, shall (notwithstanding any explicit or implicit disclosure that it may be acting on behalf of another party) be deemed for all purposes to be a party to, and bound by, the terms of the Deposit Agreement and the applicable ADR(s) to the same extent as, and as if the DTC Participant were, the Holder of such ADSs.
Section 1.20 “Exchange Act” shall mean the United States Securities Exchange Act of 1934, as amended from time to time.
Section 1.21 “Foreign Currency” shall mean any currency other than Dollars.
Section 1.22 “Full Entitlement ADR(s)”, “Full Entitlement ADS(s)” and “Full Entitlement Share(s)” shall have the respective meanings set forth in Section 2.12.
Section 1.23 “Holder(s)” shall mean the person(s) in whose name the ADSs are registered on the books of the Depositary (or the Registrar, if any) maintained for such purpose. A Holder may or may not be a Beneficial Owner. If a Holder is not the Beneficial Owner of the ADS(s) registered in its name, such person shall be deemed, for all purposes hereunder, to have all requisite authority to act on behalf of the Beneficial Owners of the ADSs registered in its name. The manner in which a Holder holds ADSs (e.g., in certificated vs. uncertificated form) may affect the rights and obligations of, and the manner in which, and the extent to which, the services are made available to, Holders pursuant to the terms of the Deposit Agreement.
Section 1.24 “Partial Entitlement ADR(s)”, “Partial Entitlement ADS(s)” and “Partial Entitlement Share(s)” shall have the respective meanings set forth in Section 2.12.
Section 1.25 “Pounds”, “Pence” and “£” shall refer to the lawful currency of England and Wales.
Section 1.26 “Principal Office” shall mean, when used with respect to the Depositary, the principal office of the Depositary at which at any particular time its depositary receipts business shall be administered, which, at the date of the Deposit Agreement, is located at 388 Greenwich Street, New York, New York 10013, U.S.A.
Section 1.27 “Registrar” shall mean the Depositary or any bank or trust company having an office in the Borough of Manhattan, The City of New York, which shall be appointed by the Depositary to register issuances, transfers and cancellations of ADSs as herein provided, and shall include any co-registrar appointed by the Depositary for such purposes. Registrars (other than the Depositary) may be removed and substitutes appointed by the Depositary. Each Registrar (other than the Depositary) appointed pursuant to the Deposit Agreement shall be required to give notice in writing to the Depositary accepting such appointment and agreeing to be bound by the applicable terms of the Deposit Agreement.
Section 1.28 “Restricted Securities” shall mean Shares, Deposited Securities or ADSs which (i) have been acquired directly or indirectly from the Company or any of its Affiliates in a transaction or chain of transactions not involving any public offering and are subject to resale limitations under the Securities Act or the rules issued thereunder, or (ii) are held by an executive officer or director (or persons performing similar functions) or other Affiliate of the Company, or (iii) are subject to other restrictions on sale or deposit under the laws of the United States, England and Wales, or under a shareholder agreement or the Articles of Association of the Company or under the regulations of an applicable securities exchange unless, in each case, such Shares, Deposited Securities or ADSs are being transferred or sold to persons other than an Affiliate of the Company in a transaction (a) covered by an effective resale registration statement, or (b) exempt from the registration requirements of the Securities Act (as hereinafter defined), and the Shares, Deposited Securities or ADSs are not, when held by such person(s), Restricted Securities.
Section 1.29 “Restricted ADR(s)”, “Restricted ADS(s)” and “Restricted Shares” shall have the respective meanings set forth in Section 2.14.
Section 1.30 “Securities Act” shall mean the United States Securities Act of 1933, as amended from time to time.
Section 1.31 “Share Registrar” shall mean Computershare Investor Services PLC or any other institution organized under the laws of England and Wales appointed by the Company from time to time to carry out the duties of registrar for the Shares, and any successor thereto.
Section 1.32 “Shares” shall mean the Company’s ordinary shares, nominal value £0.002 per share, validly issued and outstanding and fully paid and may, if the Depositary so agrees after consultation with the Company, include evidence of the right to receive Shares; provided that in no event shall Shares include evidence of the right to receive Shares with respect to which the full subscription price has not been paid or Shares as to which preemptive rights have theretofore not been validly waived or exercised; provided further, however, that, if there shall occur any change in nominal value, sub-division, consolidation, reclassification, exchange, conversion or any other event described in Section 4.11 in respect of the Shares of the Company, the term “Shares” shall thereafter, to the maximum extent permitted by law, represent the successor securities resulting from such event.
Section 1.33 “Uncertificated ADS(s)” shall have the meaning set forth in Section 2.13.
Section 1.34 “United States” and “U.S.” shall have the meaning assigned to it in Regulation S as promulgated by the Commission under the Securities Act.
ARTICLE II
APPOINTMENT OF DEPOSITARY; FORM OF RECEIPTS;
DEPOSIT OF SHARES; EXECUTION AND
DELIVERY, TRANSFER AND SURRENDER OF RECEIPTS
Section 2.1 Appointment of Depositary. The Company hereby appoints the Depositary as depositary for the Deposited Property and hereby authorizes and directs the Depositary to act in accordance with the terms and conditions set forth in the Deposit Agreement and the applicable ADRs. Each Holder and each Beneficial Owner, upon acceptance of any ADSs (or any interest therein) issued in accordance with the terms and conditions of the Deposit Agreement shall be deemed for all purposes to (a) be a party to and bound by the terms of the Deposit Agreement and the applicable ADR(s), and (b) appoint the Depositary its attorney-in-fact, with full power to delegate, to act on its behalf and to take any and all actions contemplated in the Deposit Agreement and the applicable ADR(s), to adopt any and all procedures necessary to comply with applicable law and to take such action as the Depositary in its sole discretion may deem necessary or appropriate to carry out the purposes of the Deposit Agreement and the applicable ADR(s), the taking of such actions to be the conclusive determinant of the necessity and appropriateness thereof.
Section 2.2 Form and Transferability of ADSs.
(a) Form. Certificated ADSs shall be evidenced by definitive ADRs which shall be engraved, printed, lithographed or produced in such other manner as may be agreed upon by the Company and the Depositary. ADRs may be issued under the Deposit Agreement in denominations of any whole number of ADSs. The ADRs shall be substantially in the form set forth in Exhibit A to the Deposit Agreement, with any appropriate insertions, modifications and omissions, in each case as otherwise contemplated in the Deposit Agreement or required by law. ADRs shall be (i) dated, (ii) signed by the manual or facsimile signature of a duly authorized signatory of the Depositary, (iii) countersigned by the manual or facsimile signature of a duly authorized signatory of the Registrar, and (iv) registered in the books maintained by the Registrar for the registration of issuances and transfers of ADSs. No ADR and no Certificated ADS evidenced thereby shall be entitled to any benefits under the Deposit Agreement or be valid or enforceable for any purpose against the Depositary or the Company, unless such ADR shall have been so dated, signed, countersigned and registered. ADRs bearing the facsimile signature of a duly-authorized signatory of the Depositary or the Registrar, who at the time of signature was a duly-authorized signatory of the Depositary or the Registrar, as the case may be, shall bind the Depositary, notwithstanding the fact that such signatory has ceased to be so authorized prior to the Delivery of such ADR by the Depositary. The ADRs shall bear a CUSIP number that is different from any CUSIP number that was, is or may be assigned to any depositary receipts previously or subsequently issued pursuant to any other arrangement between the Depositary (or any other depositary) and the Company and which are not ADRs outstanding hereunder.
(b) Legends. The ADRs may be endorsed with, or have incorporated in the text thereof, such legends or recitals not inconsistent with the provisions of the Deposit Agreement as may be (i) necessary to enable the Depositary and the Company to perform their respective obligations hereunder, (ii) required to comply with any applicable laws or regulations, or with the rules and regulations of any securities exchange or market upon which ADSs may be traded, listed or quoted, or to conform with any usage with respect thereto, (iii) necessary to indicate any special limitations or restrictions to which any particular ADRs or ADSs are subject by reason of the date of issuance of the Deposited Securities or otherwise, or (iv) required by any book-entry system in which the ADSs are held. Holders and Beneficial Owners shall be deemed, for all purposes, to have notice of, and to be bound by, the terms and conditions of the legends set forth, in the case of Holders, on the ADR registered in the name of the applicable Holders or, in the case of Beneficial Owners, on the ADR representing the ADSs owned by such Beneficial Owners.
(c) Title. Subject to the limitations contained herein and in the ADR, title to an ADR (and to each Certificated ADS evidenced thereby) shall be transferable upon the same terms as a certificated security under the laws of the State of New York, provided that, in the case of Certificated ADSs, such ADR has been properly endorsed or is accompanied by proper instruments of transfer. Notwithstanding any notice to the contrary, the Depositary and the Company may deem and treat the Holder of an ADS (that is, the person in whose name an ADS is registered on the books of the Depositary) as the absolute owner thereof for all purposes. Neither the Depositary nor the Company shall have any obligation nor be subject to any liability under the Deposit Agreement or any ADR to any holder or any Beneficial Owner unless, in the case of a holder of ADSs, such holder is the Holder registered on the books of the Depositary or, in the case of a Beneficial Owner, such Beneficial Owner, or the Beneficial Owner’s representative, is the Holder registered on the books of the Depositary.
(d) Book-Entry Systems. The Depositary shall make arrangements for the acceptance of the ADSs into DTC. All ADSs held through DTC will be registered in the name of the nominee for DTC (currently “Cede & Co.”). As such, the nominee for DTC will be the only “Holder” of all ADSs held through DTC. Unless issued by the Depositary as Uncertificated ADSs, the ADSs registered in the name of Cede & Co. will be evidenced by one or more ADR(s) in the form of a “Balance Certificate,” which will provide that it represents the aggregate number of ADSs from time to time indicated in the records of the Depositary as being issued hereunder and that the aggregate number of ADSs represented thereby may from time to time be increased or decreased by making adjustments on such records of the Depositary and of DTC or its nominee as hereinafter provided. Citibank, N.A. (or such other entity as is appointed by DTC or its nominee) may hold the “Balance Certificate” as custodian for DTC. Each Beneficial Owner of ADSs held through DTC must rely upon the procedures of DTC and the DTC Participants to exercise or be entitled to any rights attributable to such ADSs. The DTC Participants shall for all purposes be deemed to have all requisite power and authority to act on behalf of the Beneficial Owners of the ADSs held in the DTC Participants’ respective accounts in DTC and the Depositary shall for all purposes be authorized to rely upon any instructions and information given to it by DTC Participants. So long as ADSs are held through DTC or unless otherwise required by law, ownership of beneficial interests in the ADSs registered in the name of the nominee for DTC will be shown on, and transfers of such ownership will be effected only through, records maintained by (i) DTC or its nominee (with respect to the interests of DTC Participants), or (ii) DTC Participants or their nominees (with respect to the interests of clients of DTC Participants). Any distributions made, and any notices given, by the Depositary to DTC under the terms of the Deposit Agreement shall (unless otherwise specified by the Depositary) satisfy the Depositary’s obligations under the Deposit Agreement to make such distributions, and give such notices, in respect of the ADSs held in DTC (including, for avoidance of doubt, to the DTC Participants holding the ADSs in their DTC accounts and to the Beneficial Owners of such ADSs).
Section 2.3 Deposit of Shares. Subject to the terms and conditions of the Deposit Agreement and applicable law, Shares or evidence of rights to receive Shares (other than Restricted Securities) may be deposited by any person (including the Depositary in its individual capacity but subject, however, in the case of the Company or any Affiliate of the Company, to Section 5.7) at any time, whether or not the transfer books of the Company or the Share Registrar, if any, are closed, by Delivery of the Shares to the Custodian. Every deposit of Shares shall be accompanied by the following: (A) (i) in the case of Shares represented by certificates issued in registered form, the certificate(s) representing such Shares and, where relevant, appropriate instruments of transfer or endorsement, in a form reasonably satisfactory to the Custodian, (ii) in the case of Shares represented by certificates in bearer form, the requisite coupons and talons pertaining thereto, and (iii) in the case of Shares delivered by book-entry transfer and recordation, confirmation of such book-entry transfer and recordation in the books of the Share Registrar or of CREST, as applicable, to the Custodian or that irrevocable instructions have been given to cause such Shares to be so issued or transferred, as applicable, and recorded, (B) such certifications and payments (including, without limitation, the Depositary’s fees and related charges) and evidence of such payments (including, without limitation, stamping or otherwise marking such Shares by way of receipt) as may be reasonably required by the Depositary or the Custodian in accordance with the provisions of the Deposit Agreement and applicable law, (C) if the Depositary so requires, a written order directing the Depositary to issue and deliver to, or upon the written order of, the person(s) stated in such order the number of ADSs representing the Shares so deposited, (D) evidence reasonably satisfactory to the Depositary (which may be an opinion of counsel) that all necessary approvals have been granted by, or there has been compliance with the rules and regulations of, any applicable governmental agency in England and Wales, and (E) if the Depositary so requires, (i) an agreement, assignment or instrument reasonably satisfactory to the Depositary or the Custodian which provides for the prompt transfer by any person in whose name the Shares are or have been recorded to the Custodian of any distribution, or right to subscribe for additional Shares or to receive other property in respect of any such deposited Shares or, in lieu thereof, such indemnity or other agreement as shall be reasonably satisfactory to the Depositary or the Custodian and (ii) if the Shares are registered in the name of the person on whose behalf they are presented for deposit, a proxy or proxies entitling the Custodian to exercise voting rights in respect of the Shares for any and all purposes until the Shares so deposited are registered in the name of the Depositary, the Custodian or any nominee.
Without limiting any other provision of the Deposit Agreement, the Depositary shall instruct the Custodian not to, and the Depositary shall not knowingly, accept for deposit (a) any Restricted Securities (except as contemplated by Section 2.14) nor (b) any fractional Shares or fractional Deposited Securities nor (c) a number of Shares or Deposited Securities which upon application of the ADS to Shares ratio would give rise to fractional ADSs. No Shares shall be accepted for deposit unless accompanied by evidence, if any is required by the Depositary, that is reasonably satisfactory to the Depositary or the Custodian that all conditions to such deposit have been satisfied by the person depositing such Shares under the laws and regulations of England and Wales and any necessary approval has been granted by any applicable governmental body in England and Wales, if any. The Depositary may issue ADSs against evidence of rights to receive Shares from the Company, any agent of the Company or any custodian, registrar, transfer agent, clearing agency or other entity involved in ownership or transaction records in respect of the Shares. Such evidence of rights shall consist of written blanket or specific guarantees of ownership of Shares furnished by the Company or any such custodian, registrar, transfer agent, clearing agency or other entity involved in ownership or transaction records in respect of the Shares.
Without limitation of the foregoing, the Depositary shall not knowingly accept for deposit under the Deposit Agreement (A) any Shares or other securities required to be registered under the provisions of the Securities Act, unless (i) a registration statement is in effect as to such Shares or other securities or (ii) the deposit is made upon terms contemplated in Section 2.14, or (B) any Shares or other securities the deposit of which would violate any provisions of the Articles of Association of the Company or English Law. For purposes of the foregoing sentence, the Depositary shall be entitled to rely upon representations and warranties made or deemed made pursuant to the Deposit Agreement and shall not be required to make any further investigation. The Depositary will comply with written instructions of the Company (received by the Depositary reasonably in advance) not to accept for deposit hereunder any Shares identified in such instructions at such times and under such circumstances as may reasonably be specified in such instructions in order to facilitate the Company’s compliance with the securities laws of the United States.
Section 2.4 Registration and Safekeeping of Deposited Securities. The Depositary shall instruct the Custodian upon each Delivery of registered Shares being deposited hereunder with the Custodian (or other Deposited Securities pursuant to Article IV hereof), together with the other documents above specified, to present such Shares, together with the appropriate instrument(s) of transfer or endorsement, duly stamped, to the Share Registrar for transfer and registration of the Shares (as soon as transfer and registration can be accomplished and at the expense of the person for whom the deposit is made) in the name of the Depositary, the Custodian or a nominee of either. Deposited Securities shall be held by the Depositary, or by a Custodian for the account and to the order of the Depositary or a nominee of the Depositary, in each case, on behalf of the Holders and Beneficial Owners, at such place(s) as the Depositary or the Custodian shall determine. Notwithstanding anything else contained in the Deposit Agreement, any ADR(s), or any other instruments or agreements relating to the ADSs and the corresponding Deposited Property, the registration of the Deposited Securities in the name of the Depositary, the Custodian or any of their respective nominees, shall, to the maximum extent permitted by applicable law, vest in the Depositary, the Custodian or the applicable nominee the record ownership in the applicable Deposited Securities with the beneficial ownership rights and interests in such Deposited Securities being at all times vested with the Beneficial Owners of the ADSs representing the Deposited Securities. Notwithstanding the foregoing, the Depositary, the Custodian and the applicable nominee shall at all times be entitled to exercise the beneficial ownership rights in all Deposited Property, in each case only on behalf of the Holders and Beneficial Owners of the ADSs representing the Deposited Property, upon the terms set forth in the Deposit Agreement and, if applicable, the ADR(s) representing the ADSs. The Depositary, the Custodian and their respective nominees shall for all purposes be deemed to have all requisite power and authority to act in respect of Deposited Property on behalf of the Holders and Beneficial Owners of ADSs representing the Deposited Property, and upon making payments to, or acting upon instructions from, or information provided by, the Depositary, the Custodian or their respective nominees all persons shall be authorized to rely upon such power and authority.
Section 2.5 Issuance of ADSs. The Depositary has made arrangements with the Custodian for the Custodian to confirm to the Depositary upon receipt of a deposit of Shares (i) that a deposit of Shares has been made pursuant to Section 2.3, (ii) that such Deposited Securities have been recorded in the name of the Depositary, the Custodian or a nominee of either on the shareholders’ register maintained by or on behalf of the Company by the Share Registrar or on the books of CREST, (iii) that all required documents have been received, and (iv) the person(s) to whom or upon whose order ADSs are deliverable in respect thereof and the number of ADSs to be so delivered. Such notification may be made by letter, cable, telex, SWIFT message or, at the risk and expense of the person making the deposit, by facsimile or other means of electronic transmission. Upon receiving such notice from the Custodian, the Depositary, subject to the terms and conditions of the Deposit Agreement and applicable law, shall issue the ADSs representing the Shares so deposited to or upon the order of the person(s) named in the notice delivered to the Depositary and, if applicable, shall execute and deliver at its Principal Office Receipt(s) registered in the name(s) requested by such person(s) and evidencing the aggregate number of ADSs to which such person(s) is/are entitled, but, in each case, only upon payment to the Depositary of the charges of the Depositary for accepting a deposit of Shares and issuing ADSs (as set forth in Section 5.9 and Exhibit B hereto) and all taxes and governmental charges and fees payable in connection with such deposit and the transfer of the Shares and the issuance of the ADS(s). The Depositary shall only issue ADSs in whole numbers and deliver, if applicable, ADR(s) evidencing whole numbers of ADSs.
Section 2.6 Transfer, Combination and Split-up of ADRs.
(a) Transfer. The Registrar shall register the transfer of ADRs (and of the ADSs represented thereby) on the books maintained for such purpose and the Depositary shall (x) cancel such ADRs and execute new ADRs evidencing the same aggregate number of ADSs as those evidenced by the ADRs canceled by the Depositary, (y) cause the Registrar to countersign such new ADRs and (z) Deliver such new ADRs to or upon the order of the person entitled thereto, if each of the following conditions has been satisfied: (i) the ADRs have been duly Delivered by the Holder (or by a duly authorized attorney of the Holder) to the Depositary at its Principal Office for the purpose of effecting a transfer thereof, (ii) the surrendered ADRs have been properly endorsed or are accompanied by proper instruments of transfer (including signature guarantees in accordance with standard securities industry practice), (iii) the surrendered ADRs have been duly stamped (if required by the laws of the State of New York or of the United States), and (iv) all applicable fees and charges of, and expenses incurred by, the Depositary and all applicable taxes and governmental charges (as are set forth in Section 5.9 and Exhibit B hereto) have been paid, subject, however, in each case, to the terms and conditions of the applicable ADRs, of the Deposit Agreement and of applicable law, in each case as in effect at the time thereof.
(b) Combination & Split-Up. The Registrar shall register the split-up or combination of ADRs (and of the ADSs represented thereby) on the books maintained for such purpose and the Depositary shall (x) cancel such ADRs and execute new ADRs for the number of ADSs requested, but in the aggregate not exceeding the number of ADSs evidenced by the ADRs canceled by the Depositary, (y) cause the Registrar to countersign such new ADRs and (z) Deliver such new ADRs to or upon the order of the Holder thereof, if each of the following conditions has been satisfied: (i) the ADRs have been duly Delivered by the Holder (or by a duly authorized attorney of the Holder) to the Depositary at its Principal Office for the purpose of effecting a split-up or combination thereof, and (ii) all applicable fees and charges of, and expenses incurred by, the Depositary and all applicable taxes and governmental charges (as are set forth in Section 5.9 and Exhibit B hereto) have been paid, subject, however, in each case, to the terms and conditions of the applicable ADRs, of the Deposit Agreement and of applicable law, in each case as in effect at the time thereof.
Section 2.7 Surrender of ADSs and Withdrawal of Deposited Securities. The Holder of ADSs shall be entitled to Delivery (at the Custodian’s designated office) of the Deposited Securities at the time represented by the ADSs upon satisfaction of each of the following conditions: (i) the Holder (or a duly-authorized attorney of the Holder) has duly Delivered ADSs to the Depositary at its Principal Office (and if applicable, the ADRs evidencing such ADSs) for the purpose of withdrawal of the Deposited Securities represented thereby, (ii) if applicable and so required by the Depositary, the ADRs Delivered to the Depositary for such purpose have been properly endorsed in blank or are accompanied by proper instruments of transfer in blank (including signature guarantees in accordance with standard securities industry practice), (iii) if so required by the Depositary, the Holder of the ADSs has executed and delivered to the Depositary a written order directing the Depositary to cause the Deposited Securities being withdrawn to be Delivered to or upon the written order of the person(s) designated in such order, and (iv) all applicable fees and charges of, and expenses incurred by, the Depositary and all applicable taxes and governmental charges (as are set forth in Section 5.9 and Exhibit B) have been paid, subject, however, in each case, to the terms and conditions of the ADRs evidencing the surrendered ADSs, of the Deposit Agreement, of the Company’s Articles of Association and of any applicable laws and the rules of CREST, and to any provisions of or governing the Deposited Securities , in each case as in effect at the time thereof.
Upon satisfaction of each of the conditions specified above, the Depositary (i) shall cancel the ADSs Delivered to it (and, if applicable, the ADR(s) evidencing the ADSs so Delivered), (ii) shall direct the Registrar to record the cancellation of the ADSs so Delivered on the books maintained for such purpose, and (iii) shall direct the Custodian to Deliver, or cause the Delivery of, in each case, without unreasonable delay, the Deposited Securities represented by the ADSs so canceled together with any certificate or other document of title for the Deposited Securities, or evidence of the electronic transfer thereof (if available), as the case may be, to or upon the written order of the person(s) designated in the order delivered to the Depositary for such purpose, subject however, in each case, to the terms and conditions of the Deposit Agreement, of the ADRs evidencing the ADSs so canceled, of the Articles of Association of the Company, of any applicable laws and of the rules of CREST, and to the terms and conditions of or governing the Deposited Securities, in each case as in effect at the time thereof.
The Depositary shall not accept for surrender ADSs representing less than one (1) Share. In the case of Delivery to it of ADSs representing a number other than a whole number of Shares, the Depositary shall cause ownership of the appropriate whole number of Shares to be Delivered in accordance with the terms hereof, and shall, at the discretion of the Depositary, either (i) return to the person surrendering such ADSs the number of ADSs representing any remaining fractional Share, or (ii) sell or cause to be sold the fractional Share represented by the ADSs so surrendered and remit the proceeds of such sale (net of (a) applicable fees and charges of, and expenses incurred by, the Depositary and (b) applicable taxes required to be withheld as a result of such sale) to the person surrendering the ADSs.
Notwithstanding anything else contained in any ADR or the Deposit Agreement, the Depositary may make delivery at the Principal Office of the Depositary of Deposited Property consisting of (i) any cash dividends or cash distributions, or (ii) any proceeds from the sale of any non-cash distributions, which are at the time held by the Depositary in respect of the Deposited Securities represented by the ADSs surrendered for cancellation and withdrawal. At the request, risk and expense of any Holder so surrendering ADSs, and for the account of such Holder, the Depositary shall direct the Custodian to forward (to the extent permitted by law) any Deposited Property (other than Deposited Securities) held by the Custodian in respect of such ADSs to the Depositary for delivery at the Principal Office of the Depositary. Such direction shall be given by letter or, at the request, risk and expense of such Holder, by cable, telex or facsimile transmission.
Section 2.8 Limitations on Execution and Delivery, Transfer, etc. of ADSs; Suspension of Delivery, Transfer, etc.
(a) Additional Requirements. As a condition precedent to the execution and Delivery, the registration of issuance, transfer, split-up, combination or surrender, of any ADS, the delivery of any distribution thereon, or the withdrawal of any Deposited Property, the Depositary or the Custodian may require (i) payment from the depositor of Shares or presenter of ADSs or of an ADR of a sum sufficient to reimburse it for any tax or other governmental charge and any stock transfer or registration fee with respect thereto (including any such tax or charge and fee with respect to Shares being deposited or withdrawn) and payment of any applicable fees and charges of the Depositary as provided in Section 5.9 and Exhibit B, (ii) the production of proof reasonably satisfactory to it as to the identity and genuineness of any signature or any other matter contemplated by Section 3.1, and (iii) compliance with (A) any laws or governmental regulations relating to the execution and Delivery of ADRs or ADSs or to the withdrawal of Deposited Securities and (B) such reasonable regulations as the Depositary and the Company may establish consistent with the provisions of the representative ADR, if applicable, the Deposit Agreement and applicable law.
(b) Additional Limitations. The issuance of ADSs against deposits of Shares generally or against deposits of particular Shares may be suspended, or the deposit of particular Shares may be refused, or the registration of transfer of ADSs in particular instances may be refused, or the registration of transfers of ADSs generally may be suspended, during any period when the transfer books of the Company, the Depositary, a Registrar or the Share Registrar are closed or if any such action is deemed necessary or advisable by the Depositary or the Company, in good faith, at any time or from time to time because of any requirement of law or regulation, any government or governmental body or commission or any securities exchange on which the ADSs or Shares are listed, or under any provision of the Deposit Agreement or the representative ADR(s), if applicable, or under any provision of, or governing, the Deposited Securities, or because of a meeting of shareholders of the Company or for any other reason, subject, in all cases, to Section 7.8(a).
(c) Regulatory Restrictions. Notwithstanding any provision of the Deposit Agreement or any ADR(s) to the contrary, Holders are entitled to surrender outstanding ADSs to withdraw the Deposited Securities associated herewith at any time subject only to (i) temporary delays caused by closing the transfer books of the Depositary or the Company or the deposit of Shares in connection with voting at a shareholders’ meeting or the payment of dividends, (ii) the payment of fees, taxes and similar charges, (iii) compliance with any U.S. or foreign laws or governmental regulations relating to the ADSs or to the withdrawal of the Deposited Securities, and (iv) other circumstances specifically contemplated by Instruction I.A.(l) of the General Instructions to Form F-6 (as such General Instructions may be amended from time to time).
Section 2.9 Lost ADRs, etc. In case any ADR shall be mutilated, destroyed, lost, or stolen, the Depositary shall execute and deliver a new ADR of like tenor at the expense of the Holder (a) in the case of a mutilated ADR, in exchange of and substitution for such mutilated ADR upon cancellation thereof, or (b) in the case of a destroyed, lost or stolen ADR, in lieu of and in substitution for such destroyed, lost, or stolen ADR, after the Holder thereof (i) has submitted to the Depositary a written request for such exchange and substitution before the Depositary has notice that the ADR has been acquired by a bona fide purchaser, (ii) has provided such security or indemnity (including an indemnity bond) as may be required by the Depositary to save it and any of its agents harmless, and (iii) has satisfied any other reasonable requirements imposed by the Depositary, including, without limitation, evidence satisfactory to the Depositary of such destruction, loss or theft of such ADR, the authenticity thereof and the Holder’s ownership thereof.
Section 2.10 Cancellation and Destruction of Surrendered ADRs; Maintenance of Records. All ADRs surrendered to the Depositary shall be canceled by the Depositary. Canceled ADRs shall not be entitled to any benefits under the Deposit Agreement or be valid or enforceable against the Depositary for any purpose. The Depositary is authorized to destroy ADRs so canceled, provided the Depositary maintains a record of all destroyed ADRs. Any ADSs held in book-entry form (e.g., through accounts at DTC) shall be deemed canceled when the Depositary causes the number of ADSs evidenced by the Balance Certificate to be reduced by the number of ADSs surrendered (without the need to physically destroy the Balance Certificate).
Section 2.11 Escheatment. In the event any unclaimed property relating to the ADSs, for any reason, is in the possession of Depositary and has not been claimed by the Holder thereof or cannot be delivered to the Holder thereof through usual channels, the Depositary shall, upon expiration of any applicable statutory period relating to abandoned property laws, escheat such unclaimed property to the relevant authorities in accordance with the laws of each of the relevant States of the United States.
Section 2.12 Partial Entitlement ADSs. In the event any Shares are deposited which (i) entitle the holders thereof to receive a per-share distribution or other entitlement in an amount different from the Shares then on deposit or (ii) are not fully fungible (including, without limitation, as to settlement or trading) with the Shares then on deposit (the Shares then on deposit collectively, “Full Entitlement Shares” and the Shares with different entitlement, “Partial Entitlement Shares”), the Depositary shall (i) cause the Custodian to hold Partial Entitlement Shares separate and distinct from Full Entitlement Shares, and (ii) subject to the terms of the Deposit Agreement, issue ADSs representing Partial Entitlement Shares which are separate and distinct from the ADSs representing Full Entitlement Shares, by means of separate CUSIP numbering and legending (if necessary) and, if applicable, by issuing ADRs evidencing such ADSs with applicable notations thereon (“Partial Entitlement ADSs/ADRs” and “Full Entitlement ADSs/ADRs”, respectively). If and when Partial Entitlement Shares become Full Entitlement Shares, the Depositary shall (a) give notice thereof to Holders of Partial Entitlement ADSs and give Holders of Partial Entitlement ADRs the opportunity to exchange such Partial Entitlement ADRs for Full Entitlement ADRs, (b) cause the Custodian to transfer the Partial Entitlement Shares into the account of the Full Entitlement Shares, and (c) take such actions as are necessary to remove the distinctions between (i) the Partial Entitlement ADRs and ADSs, on the one hand, and (ii) the Full Entitlement ADRs and ADSs on the other. Holders and Beneficial Owners of Partial Entitlement ADSs shall only be entitled to the entitlements of Partial Entitlement Shares. Holders and Beneficial Owners of Full Entitlement ADSs shall be entitled only to the entitlements of Full Entitlement Shares. All provisions and conditions of the Deposit Agreement shall apply to Partial Entitlement ADRs and ADSs to the same extent as Full Entitlement ADRs and ADSs, except as contemplated by this Section 2.12. The Depositary is authorized to take any and all other actions as may be necessary (including, without limitation, making the necessary notations on ADRs) to give effect to the terms of this Section 2.12. The Company agrees to give timely written notice to the Depositary if any Shares issued or to be issued are Partial Entitlement Shares and shall assist the Depositary with the establishment of procedures enabling the identification of Partial Entitlement Shares upon Delivery to the Custodian.
Section 2.13 Certificated/Uncertificated ADSs. Notwithstanding any other provision of the Deposit Agreement, the Depositary may, at any time and from time to time, issue ADSs that are not evidenced by ADRs (such ADSs, the “Uncertificated ADS(s)” and the ADS(s) evidenced by ADR(s), the “Certificated ADS(s)”). When issuing and maintaining Uncertificated ADS(s) under the Deposit Agreement, the Depositary shall at all times be subject to (i) the standards applicable to registrars and transfer agents maintaining direct registration systems for equity securities in New York and issuing uncertificated securities under New York law, and (ii) the terms of New York law applicable to uncertificated equity securities. Uncertificated ADSs shall not be represented by any instruments but shall be evidenced by registration in the books of the Depositary maintained for such purpose. Holders of Uncertificated ADSs, that are not subject to any registered pledges, liens, restrictions or adverse claims of which the Depositary has notice at such time, shall at all times have the right to exchange the Uncertificated ADS(s) for Certificated ADS(s) of the same type and class, subject in each case to (x) applicable laws and any rules and regulations the Depositary may have established in respect of the Uncertificated ADSs, and (y) the continued availability of Certificated ADSs in the U.S. Holders of Certificated ADSs shall, if the Depositary maintains a direct registration system for the ADSs, have the right to exchange the Certificated ADSs for Uncertificated ADSs upon (i) the due surrender of the Certificated ADS(s) to the Depositary for such purpose and (ii) the presentation of a written request to that effect to the Depositary, subject in each case to (a) all liens and restrictions noted on the ADR evidencing the Certificated ADS(s) and all adverse claims of which the Depositary then has notice, (b) the terms of the Deposit Agreement and the rules and regulations that the Depositary may establish for such purposes hereunder, (c) applicable law, and (d) payment of the Depositary fees and expenses applicable to such exchange of Certificated ADS(s) for Uncertificated ADS(s). Uncertificated ADSs shall in all material respects be identical to Certificated ADS(s) of the same type and class, except that (i) no ADR(s) shall be, or shall need to be, issued to evidence Uncertificated ADS(s), (ii) Uncertificated ADS(s) shall, subject to the terms of the Deposit Agreement, be transferable upon the same terms and conditions as uncertificated securities under New York law, (iii) the ownership of Uncertificated ADS(s) shall be recorded on the books of the Depositary maintained for such purpose and evidence of such ownership shall be reflected in periodic statements provided by the Depositary to the Holder(s) in accordance with applicable New York law, (iv) the Depositary may from time to time, upon notice to the Holders of Uncertificated ADSs affected thereby, establish rules and regulations, and amend or supplement existing rules and regulations, as may be deemed reasonably necessary to maintain Uncertificated ADS(s) on behalf of Holders, provided that (a) such rules and regulations do not conflict with the terms of the Deposit Agreement and applicable law, and (b) the terms of such rules and regulations are readily available to Holders upon request, (v) the Uncertificated ADS(s) shall not be entitled to any benefits under the Deposit Agreement or be valid or enforceable for any purpose against the Depositary or the Company unless such Uncertificated ADS(s) is/are registered on the books of the Depositary maintained for such purpose, (vi) the Depositary may, in connection with any deposit of Shares resulting in the issuance of Uncertificated ADSs and with any transfer, pledge, release and cancellation of Uncertificated ADSs, require the prior receipt of such documentation as the Depositary may deem reasonably appropriate, and (vii) upon termination of the Deposit Agreement, the Depositary shall not require Holders of Uncertificated ADSs to affirmatively instruct the Depositary before remitting proceeds from the sale of the Deposited Property represented by such Holders' Uncertificated ADSs under the terms of Section 6.2. When issuing ADSs under the terms of the Deposit Agreement, including, without limitation, issuances pursuant to Sections 2.5, 4.2, 4.3, 4.4, 4.5 and 4.11, the Depositary may in its discretion determine to issue Uncertificated ADSs rather than Certificated ADSs, unless otherwise specifically instructed by the applicable Holder to issue Certificated ADSs. All provisions and conditions of the Deposit Agreement shall apply to Uncertificated ADSs to the same extent as to Certificated ADSs, except as contemplated by this Section 2.13. The Depositary is authorized and directed to take any and all actions and establish any and all procedures deemed reasonably necessary to give effect to the terms of this Section 2.13. Any references in the Deposit Agreement or any ADR(s) to the terms “American Depositary Share(s)” or “ADS(s)” shall, unless the context otherwise requires, include Certificated ADS(s) and Uncertificated ADS(s). Except as set forth in this Section 2.13 and except as required by applicable law, the Uncertificated ADSs shall be treated as ADSs issued and outstanding under the terms of the Deposit Agreement. In the event that, in determining the rights and obligations of parties hereto with respect to any Uncertificated ADSs, any conflict arises between (a) the terms of the Deposit Agreement (other than this Section 2.13) and (b) the terms of this Section 2.13, the terms and conditions set forth in this Section 2.13 shall be controlling and shall govern the rights and obligations of the parties to the Deposit Agreement pertaining to the Uncertificated ADSs.
Section 2.14 Restricted ADSs. The Depositary shall, at the request and expense of the Company, establish procedures enabling the deposit hereunder of Shares that are Restricted Securities in order to enable the holder of such Shares to hold its ownership interests in such Restricted Securities in the form of ADSs issued under the terms hereof (such Shares, “Restricted Shares”). Upon receipt of a written request from the Company to accept Restricted Shares for deposit hereunder, the Depositary agrees to establish procedures permitting the deposit of such Restricted Shares and the issuance of ADSs representing the right to receive, subject to the terms of the Deposit Agreement and the applicable ADR (if issued as a Certificated ADS), such deposited Restricted Shares (such ADSs, the “Restricted ADSs,” and the ADRs evidencing such Restricted ADSs, the “Restricted ADRs”). Notwithstanding anything contained in this Section 2.14, the Depositary and the Company may, to the extent not prohibited by law, agree to issue the Restricted ADSs in uncertificated form (“Uncertificated Restricted ADSs”) upon such terms and conditions as the Company and the Depositary may deem necessary and appropriate. The Company shall assist the Depositary in the establishment of such procedures and agrees that it shall take all steps necessary and satisfactory to the Depositary to ensure that the establishment of such procedures does not violate the provisions of the Securities Act or any other applicable laws. The depositors of such Restricted Shares and the Holders of the Restricted ADSs may be required prior to the deposit of such Restricted Shares, the transfer of the Restricted ADRs and Restricted ADSs or the withdrawal of the Restricted Shares represented by Restricted ADSs to provide such written certifications or agreements as the Depositary or the Company may require. The Company shall provide to the Depositary in writing the legend(s) to be affixed to the Restricted ADRs (if the Restricted ADSs are to be issued as Certificated ADSs), or to be included in the statements issued from time to time to Holders of Uncertificated ADSs (if issued as Uncertificated Restricted ADSs), which legends shall (i) be in a form reasonably satisfactory to the Depositary and (ii) contain the specific circumstances under which the Restricted ADSs, and, if applicable, the Restricted ADRs evidencing the Restricted ADSs, may be transferred or the Restricted Shares withdrawn. The Restricted ADSs issued upon the deposit of Restricted Shares shall be separately identified on the books of the Depositary and the Restricted Shares so deposited shall, to the extent required by law, be held separate and distinct from the other Deposited Securities held hereunder. The Restricted ADSs shall not be eligible for inclusion in any book-entry settlement system, including, without limitation, DTC (unless (x) otherwise agreed by the Company and the Depositary, (y) the inclusion of Restricted ADSs is acceptable to the applicable clearing system, and (z) the terms of such inclusion are generally accepted by the Commission for Restricted Securities of that type), and shall not in any way be fungible with the ADSs issued under the terms hereof that are not Restricted ADSs. The Restricted ADSs, and, if applicable, the Restricted ADRs evidencing the Restricted ADSs, shall be transferable only by the Holder thereof upon delivery to the Depositary of (i) all documentation otherwise contemplated by the Deposit Agreement and (ii) an opinion of counsel satisfactory to the Depositary setting forth, inter alia, the conditions upon which the Restricted ADSs presented, and, if applicable, the Restricted ADRs evidencing the Restricted ADSs, are transferable by the Holder thereof under applicable securities laws and the transfer restrictions contained in the legend applicable to the Restricted ADSs presented for transfer. Except as set forth in this Section 2.14 and except as required by applicable law, the Restricted ADSs and the Restricted ADRs evidencing Restricted ADSs shall be treated as ADSs and ADRs issued and outstanding under the terms of the Deposit Agreement. In the event that, in determining the rights and obligations of parties hereto with respect to any Restricted ADSs, any conflict arises between (a) the terms of the Deposit Agreement (other than this Section 2.14) and (b) the terms of (i) this Section 2.14 or (ii) the applicable Restricted ADR, the terms and conditions set forth in this Section 2.14 and of the Restricted ADR shall be controlling and shall govern the rights and obligations of the parties to the Deposit Agreement pertaining to the deposited Restricted Shares, the Restricted ADSs and Restricted ADRs.
If the Restricted ADRs, the Restricted ADSs and the Restricted Shares cease to be Restricted Securities, the Depositary, upon receipt of (x) an opinion of counsel satisfactory to the Depositary setting forth, inter alia, that the Restricted ADRs, the Restricted ADSs and the Restricted Shares are not as of such time Restricted Securities, and (y) instructions from the Company to remove the restrictions applicable to the Restricted ADRs, the Restricted ADSs and the Restricted Shares, shall (i) eliminate the distinctions and separations that may have been established between the applicable Restricted Shares held on deposit under this Section 2.14 and the other Shares held on deposit under the terms of the Deposit Agreement that are not Restricted Shares, (ii) treat the newly unrestricted ADRs and ADSs on the same terms as, and fully fungible with, the other ADRs and ADSs issued and outstanding under the terms of the Deposit Agreement that are not Restricted ADRs or Restricted ADSs, and (iii) take all actions necessary to remove any distinctions, limitations and restrictions previously existing under this Section 2.14 between the applicable Restricted ADRs and Restricted ADSs, respectively, on the one hand, and the other ADRs and ADSs that are not Restricted ADRs or Restricted ADSs, respectively, on the other hand, including, without limitation, by making the newly-unrestricted ADSs eligible for inclusion in the applicable book-entry settlement systems.
ARTICLE III
CERTAIN OBLIGATIONS OF HOLDERS
AND BENEFICIAL OWNERS OF ADSs
Section 3.1 Proofs, Certificates and Other Information. Any person presenting Shares for deposit, any Holder and any Beneficial Owner may be required, and every Holder and Beneficial Owner agrees, from time to time to provide to the Depositary and the Custodian such proof of citizenship or residence, taxpayer status, payment of all applicable taxes or other governmental charges, exchange control approval, legal or beneficial ownership of ADSs and Deposited Property, compliance with applicable laws, the terms of the Deposit Agreement or the ADR(s) evidencing the ADSs and the provisions of, or governing, the Deposited Property, to execute such certifications and to make such representations and warranties, and to provide such other information and documentation (or, in the case of Shares in registered form presented for deposit, such information relating to the registration on the books of the Company or of the Share Registrar) as the Depositary or the Custodian may deem necessary or proper or as the Company may reasonably require by written request to the Depositary consistent with its obligations under the Deposit Agreement and the applicable ADR(s). The Depositary and the Registrar, as applicable, may, and at the reasonable request of the Company, shall, to the extent practicable, withhold the execution or delivery or registration of transfer of any ADR or ADS or the distribution or sale of any dividend or distribution of rights or of the proceeds thereof or, to the extent not limited by the terms of Section 7.8(a), the delivery of any Deposited Property until such proof or other information is filed or such certifications are executed, or such representations and warranties are made, or such other documentation or information provided, in each case to the Depositary’s, the Registrar’s and the Company’s satisfaction. The Depositary shall provide the Company, in a timely manner, with copies or originals if necessary and appropriate of (i) any such proofs of citizenship or residence, taxpayer status, or exchange control approval or copies of written representations and warranties which it receives from Holders and Beneficial Owners, and (ii) any other information or documents which the Company may reasonably request and which the Depositary shall request and receive from any Holder or Beneficial Owner or any person presenting Shares for deposit or ADSs for cancellation, transfer or withdrawal. Nothing herein shall obligate the Depositary to (i) obtain any information for the Company if not provided by the Holders or Beneficial Owners, or (ii) verify or vouch for the accuracy of the information so provided by the Holders or Beneficial Owners.
Section 3.2 Liability for Taxes and Other Charges. Any tax or other governmental charge payable by the Custodian or by the Depositary with respect to any Deposited Property, ADSs or ADRs shall be payable by the Holders and Beneficial Owners to the Depositary. The Company, the Custodian and/or the Depositary may withhold or deduct from any distributions made in respect of Deposited Property held on behalf of such Holder and/or Beneficial Owner, and may sell for the account of a Holder and/or Beneficial Owner any or all of such Deposited Property and apply such distributions and sale proceeds in payment of, any taxes (including applicable interest and penalties) or charges that are or may be payable by Holders or Beneficial Owners in respect of the ADSs, Deposited Property and ADRs, the Holder and the Beneficial Owner remaining liable for any deficiency. The Custodian may refuse the deposit of Shares and the Depositary may refuse to issue ADSs, to deliver ADRs, register the transfer of ADSs, register the split-up or combination of ADRs and (subject to Section 7.8(a)) the withdrawal of Deposited Property until payment in full of such tax, charge, penalty or interest is received. Every Holder and Beneficial Owner agrees to indemnify the Depositary, the Company, the Custodian, and any of their agents, officers, employees and Affiliates for, and to hold each of them harmless from, any claims with respect to taxes (including applicable interest and penalties thereon) arising from (i) any ADSs held by such Holder and/or owned by such Beneficial Owner, (ii) the Deposited Property represented by the ADSs, and (iii) any transaction entered into by such Holder and/or Beneficial Owner in respect of the ADSs and/or the Deposited Property represented thereby. Notwithstanding anything to the contrary contained in the Deposit Agreement or any ADR, the obligations of Holders and Beneficial Owners under this Section 3.2 shall survive any transfer of ADSs, any cancellation of ADSs and withdrawal of Deposited Securities, and the termination of the Deposit Agreement.
Section 3.3 Representations and Warranties on Deposit of Shares. Each person depositing Shares under the Deposit Agreement shall be deemed thereby to represent and warrant that (i) such Shares and the certificates therefor are duly authorized, validly issued, fully paid, non-assessable (i.e., not subject to call for payment of further capital) and legally obtained by such person, (ii) all preemptive (and similar) rights, if any, with respect to such Shares have been validly waived or exercised, (iii) the person making such deposit is duly authorized so to do, (iv) the Shares presented for deposit are free and clear of any lien, encumbrance, security interest, charge, mortgage or adverse claim, (v) the Shares presented for deposit are not, and the ADSs issuable upon such deposit will not be, Restricted Securities (except as contemplated in Section 2.14), (vi) the Shares presented for deposit have not been stripped of any rights or entitlements and (vii) the deposit of the Shares does not violate any applicable provisions of English law. Such representations and warranties shall survive the deposit and withdrawal of Shares, the issuance and cancellation of ADSs in respect thereof and the transfer of such ADSs. If any such representations or warranties are false in any way, the Company and the Depositary shall be authorized, at the cost and expense of the person depositing Shares, to take any and all actions necessary to correct the consequences thereof.
Section 3.4 Compliance with Information Requests. Notwithstanding any other provision of the Deposit Agreement or any ADR(s), each Holder and Beneficial Owner agrees to comply with requests from the Company pursuant to applicable law, the rules and requirements of any stock exchange on which the Shares or ADSs are, or will be, registered, traded or listed and/or the Articles of Association of the Company, which are made to provide information, inter alia, as to the capacity in which such Holder or Beneficial Owner owns ADSs (and Shares as the case may be) and regarding the identity of any other person(s) interested in such ADSs and the nature of such interest and various other matters, whether or not they are Holders and/or Beneficial Owners at the time of such request. The Depositary agrees to use its reasonable efforts to forward, upon the request of the Company and at the Company’s expense, any such request from the Company to the Holders and to forward to the Company, as promptly as practicable, any such responses to such requests received by the Depositary.
Section 3.5 Ownership Restrictions. Notwithstanding any other provision contained in the Deposit Agreement or any ADR(s) to the contrary, the Company may restrict transfers of the Shares where such transfer might result in ownership of Shares exceeding limits imposed by applicable law or the Articles of Association of the Company. The Company may also restrict, in such manner as it deems appropriate, transfers of the ADSs where such transfer may result in the total number of Shares represented by the ADSs owned by a single Holder or Beneficial Owner to exceed any such limits. The Company may, in its sole discretion but subject to applicable law, instruct the Depositary to take action with respect to the ownership interest of any Holder or Beneficial Owner in excess of the limits set forth in the preceding sentence, including, but not limited to, the imposition of restrictions on the transfer of ADSs, the removal or limitation of voting rights or mandatory sale or disposition on behalf of a Holder or Beneficial Owner of the Shares represented by the ADSs held by such Holder or Beneficial Owner in excess of such limitations, if and to the extent such disposition is permitted by applicable law and the Articles of Association of the Company. Nothing herein shall be interpreted as obligating the Depositary or the Company to ensure compliance with the ownership restrictions described in this Section 3.5.
Notwithstanding any provision of the Deposit Agreement or of the ADRs and without limiting the foregoing, by being a Holder or Beneficial Owner of an ADS, each such Holder or Beneficial Owner agrees to provide such information as the Company may request in a disclosure notice (a “Disclosure Notice”) given pursuant to the U.K. Companies Act 2006 (as amended from time to time and including any statutory modification or re-enactment thereof, the “Companies Act”) or the Articles of Association of the Company. By accepting or holding an ADS, each Holder and Beneficial Owner acknowledges that it understands that failure to comply with a Disclosure Notice may result in the imposition of sanctions against the holder of the Shares in respect of which the non-complying person is or was, or appears to be or has been, interested as provided in the Companies Act and the Articles of Association which currently include, the withdrawal of the voting rights of such Shares and (where the relevant Shares represent at least 0.25% in nominal value of the issued shares of their class (calculated exclusive of any shares held as treasury shares)) the imposition of restrictions on the rights to receive dividends on and to transfer such Shares. The Company reserves the right to instruct Holders and Beneficial Owners to deliver their ADSs for cancellation and withdrawal of the Deposited Securities so as to permit the Company to deal directly with the Holder and Beneficial Owner thereof as a holder of Shares and Holders agree to comply with such instructions. The Depositary agrees to cooperate with the Company in its efforts to inform Holders and Beneficial Owners of the Company’s exercise of its rights under this paragraph and agrees to consult with, and provide reasonable assistance without risk, liability or expense on the part of the Depositary, to the Company on the manner or manners in which it may enforce such rights with respect to any Holder or Beneficial Owner.
Section 3.6 Reporting Obligations and Regulatory Approvals. Applicable laws and regulations may require holders and beneficial owners of Shares, including the Holders and Beneficial Owners of ADSs, to satisfy reporting requirements and obtain regulatory approvals in certain circumstances. Holders and Beneficial Owners of ADSs are solely responsible for determining and complying with such reporting requirements and obtaining such approvals. Each Holder and each Beneficial Owner hereby agrees to make such determination, file such reports, and obtain such approvals to the extent and in the form required by applicable laws and regulations as in effect from time to time. Neither the Depositary, the Custodian, the Company or any of their respective agents or Affiliates shall be required to take any actions whatsoever on behalf of Holders or Beneficial Owners to determine or satisfy such reporting requirements or obtain such regulatory approvals under applicable laws and regulations.
ARTICLE IV
THE DEPOSITED SECURITIES
Section 4.1 Cash Distributions. Whenever the Company intends to make a distribution of a cash dividend or other cash distribution in respect of any Deposited Securities, the Company shall give notice thereof to the Depositary at least twenty (20) days (or such other number of days as mutually agreed to in writing by the Depositary and the Company) prior to the proposed distribution specifying, inter alia, the record date applicable for determining the holders of Deposited Securities entitled to receive such distribution. Upon the timely receipt of such notice, the Depositary shall establish the ADS Record Date upon the terms described in Section 4.9. Upon receipt of confirmation of the receipt of (x) any cash dividend or other cash distribution on any Deposited Securities, or (y) proceeds from the sale of any Deposited Property held in respect of the ADSs under the terms hereof, the Depositary will (i) if at the time of receipt thereof any amounts are received in a Foreign Currency can, in the judgment of the Depositary (pursuant to Section 4.8), be converted on a practical basis into Dollars transferable to the United States, promptly convert or cause to be converted such cash dividend, distribution or proceeds into Dollars (subject to the terms and conditions of Section 4.8), (ii) if applicable and unless previously established, establish the ADS Record Date upon the terms described in Section 4.9, and (iii) distribute promptly the amount thus received (net of (a) the applicable fees and charges of, and expenses incurred by, the Depositary, and (b) applicable taxes required to be withheld in connection with the distribution) to the Holders entitled thereto as of the ADS Record Date in proportion to the number of ADSs held as of the ADS Record Date. The Depositary shall distribute only such amount, however, as can be distributed without attributing to any Holder a fraction of one cent, and any balance not so distributed shall be held by the Depositary (without liability for interest thereon) and shall be added to and become part of the next sum received by the Depositary for distribution to Holders of ADSs outstanding at the time of the next distribution. If the Company, the Custodian or the Depositary is required to withhold and does withhold from any cash dividend or other cash distribution in respect of any Deposited Securities, or from any cash proceeds from the sales of Deposited Property, an amount on account of taxes, duties or other governmental charges, the amount distributed to Holders on the ADSs shall be reduced accordingly. Such withheld amounts shall be forwarded by the Company, the Custodian or the Depositary to the relevant governmental authority. Evidence of payment thereof by the Company shall be forwarded by the Company to the Depositary upon request. The Depositary will hold any cash amounts it is unable to distribute in a non-interest bearing account for the benefit of the applicable Holders and Beneficial Owners of ADSs until the distribution can be effected or the funds that the Depositary holds must be escheated as unclaimed property in accordance with the laws of the relevant states of the United States. Notwithstanding anything contained in the Deposit Agreement to the contrary, in the event the Company fails to give the Depositary timely notice of the proposed distribution provided for in this Section 4.1, the Depositary agrees to use commercially reasonable efforts to perform the actions contemplated in this Section 4.1, and the Company, the Holders and the Beneficial Owners acknowledge that the Depositary shall have no liability for the Depositary’s failure to perform the actions contemplated in this Section 4.1 where such notice has not been so timely given, other than its failure to use commercially reasonable efforts, as provided herein.
Section 4.2 Distribution in Shares. Whenever the Company intends to make a distribution that consists of a dividend in, or free distribution of, Shares, the Company shall give notice thereof to the Depositary at least twenty (20) days (or such other number of days as mutually agreed to in writing by the Depositary and the Company) prior to the proposed distribution, specifying, inter alia, the record date applicable to holders of Deposited Securities entitled to receive such distribution. Upon the timely receipt of such notice from the Company, the Depositary shall establish the ADS Record Date upon the terms described in Section 4.9. Upon receipt of confirmation from the Custodian of the receipt of the Shares so distributed by the Company, the Depositary shall either (i) subject to Section 5.9, distribute to the Holders as of the ADS Record Date in proportion to the number of ADSs held as of the ADS Record Date, additional ADSs, which represent in the aggregate the number of Shares received as such dividend, or free distribution, subject to the other terms of the Deposit Agreement (including, without limitation, (a) the applicable fees and charges of, and expenses incurred by, the Depositary and (b) applicable taxes required to be withheld), or (ii) if additional ADSs are not so distributed, take all actions necessary so that each ADS issued and outstanding after the ADS Record Date shall, to the extent permissible by law, thenceforth also represent rights and interests in the additional integral number of Shares distributed upon the Deposited Securities represented thereby (net of (a) the applicable fees and charges of, and expenses incurred by, the Depositary and (b) taxes). In lieu of delivering fractional ADSs, the Depositary shall sell the number of Shares or ADSs, as the case may be, represented by the aggregate of such fractions and distribute the net proceeds upon the terms described in Section 4.1. In the event that the Depositary determines that any distribution in property (including Shares) is subject to any tax or other governmental charges which the Depositary is obligated to withhold, or, if the Company in the fulfillment of its obligation under Section 5.7, has furnished an opinion of U.S. counsel determining that Shares must be registered under the Securities Act or other laws in order to be distributed to Holders (and no such registration statement has been declared effective), the Depositary may dispose of all or a portion of such property (including Shares and rights to subscribe therefor) in such amounts and in such manner, including by public or private sale, as the Depositary deems necessary and practicable, and the Depositary shall distribute the net proceeds of any such sale (after deduction of (a) applicable taxes required to be withheld and (b) fees and charges of, and expenses incurred by, the Depositary) to Holders entitled thereto upon the terms described in Section 4.1. The Depositary shall hold and/or distribute any unsold balance of such property in accordance with the provisions of the Deposit Agreement. Notwithstanding anything contained in the Deposit Agreement to the contrary, in the event the Company fails to give the Depositary timely notice of the proposed distribution provided for in this Section 4.2, the Depositary agrees to use commercially reasonable efforts to perform the actions contemplated in this Section 4.2, and the Company, the Holders and the Beneficial Owners acknowledge that the Depositary shall have no liability for the Depositary’s failure to perform the actions contemplated in this Section 4.2 where such notice has not been so timely given, other than its failure to use commercially reasonable efforts, as provided herein.
Section 4.3 Elective Distributions in Cash or Shares. Whenever the Company intends to make a distribution payable at the election of the holders of Deposited Securities in cash or in additional Shares, the Company shall give notice thereof to the Depositary at least sixty (60) days (or such other number of days as mutually agreed to in writing by the Depositary and the Company) prior to the proposed distribution specifying, inter alia, the record date applicable to holders of Deposited Securities entitled to receive such elective distribution and whether or not it wishes such elective distribution to be made available to Holders of ADSs. Upon the timely receipt of a notice indicating that the Company wishes such elective distribution to be made available to Holders of ADSs, the Depositary shall consult with the Company to determine, and the Company shall assist the Depositary in its determination, whether it is lawful and reasonably practicable to make such elective distribution available to the Holders of ADSs. The Depositary shall make such elective distribution available to Holders only if (i) the Company shall have timely requested that the elective distribution be made available to Holders, (ii) the Depositary shall have determined that such distribution is reasonably practicable and (iii) the Depositary shall have received satisfactory documentation within the terms of Section 5.7. If the above conditions are not satisfied or if the Company requests such elective distribution not to be made available to Holders of ADSs, the Depositary shall establish the ADS Record Date on the terms described in Section 4.9 and, to the extent permitted by law, distribute to the Holders, on the basis of the same determination as is made in England and Wales in respect of the Shares for which no election is made, either (X) cash upon the terms described in Section 4.1 or (Y) additional ADSs representing such additional Shares upon the terms described in Section 4.2. If the above conditions are satisfied, the Depositary shall establish an ADS Record Date on the terms described in Section 4.9 and establish procedures to enable Holders to elect the receipt of the proposed distribution in cash or in additional ADSs. The Company shall assist the Depositary in establishing such procedures to the extent necessary. If a Holder elects to receive the proposed distribution (X) in cash, the distribution shall be made upon the terms described in Section 4.1, or (Y) in ADSs, the distribution shall be made upon the terms described in Section 4.2. Nothing herein shall obligate the Depositary to make available to Holders a method to receive the elective distribution in Shares (rather than ADSs). There can be no assurance that Holders generally, or any Holder in particular, will be given the opportunity to receive elective distributions on the same terms and conditions as the holders of Shares. Notwithstanding anything contained in the Deposit Agreement to the contrary, in the event the Company fails to give the Depositary timely notice of the proposed distribution provided for in this Section 4.3, the Depositary agrees to use commercially reasonable efforts to perform the actions contemplated in this Section 4.3, and the Company, the Holders and the Beneficial Owners acknowledge that the Depositary shall have no liability for the Depositary’s failure to perform the actions contemplated in this Section 4.3 where such notice has not been so timely given, other than its failure to use commercially reasonable efforts, as provided herein.
Section 4.4 Distribution of Rights to Purchase Additional ADSs.
(a) Distribution to ADS Holders. Whenever the Company intends to distribute to the holders of the Deposited Securities rights to subscribe for additional Shares, the Company shall give notice thereof to the Depositary at least sixty (60) days (or such other number of days as mutually agreed to in writing by the Depositary and the Company) prior to the proposed distribution specifying, inter alia, the record date applicable to holders of Deposited Securities entitled to receive such distribution and whether or not it wishes such rights to be made available to Holders of ADSs. Upon the timely receipt of a notice indicating that the Company wishes such rights to be made available to Holders of ADSs, the Depositary shall consult with the Company to determine, and the Company shall assist the Depositary in its determination, whether it is lawful and reasonably practicable to make such rights available to the Holders. The Depositary shall make such rights available to Holders only if (i) the Company shall have timely requested that such rights be made available to Holders, (ii) the Depositary shall have received satisfactory documentation within the terms of Section 5.7, and (iii) the Depositary shall have determined that such distribution of rights is reasonably practicable. In the event any of the conditions set forth above are not satisfied or if the Company requests that the rights not be made available to Holders of ADSs, the Depositary shall proceed with the sale of the rights as contemplated in Section 4.4(b) below. In the event all conditions set forth above are satisfied, the Depositary shall establish the ADS Record Date (upon the terms described in Section 4.9) and establish procedures to (x) distribute rights to purchase additional ADSs (by means of warrants or otherwise), (y) enable the Holders to exercise such rights (upon payment of the subscription price and of the applicable (a) fees and charges of, and expenses incurred by, the Depositary and (b) taxes), and (z) deliver ADSs upon the valid exercise of such rights. The Company shall assist the Depositary to the extent necessary in establishing such procedures. Nothing herein shall obligate the Depositary to make available to the Holders a method to exercise rights to subscribe for Shares (rather than ADSs).
(b) Sale of Rights. If (i) the Company does not timely request the Depositary to make the rights available to Holders or requests that the rights not be made available to Holders, (ii) the Depositary fails to receive satisfactory documentation within the terms of Section 5.7, or determines it is not reasonably practicable to make the rights available to Holders, or (iii) any rights made available are not exercised and appear to be about to lapse, the Depositary shall determine whether it is lawful and reasonably practicable to sell such rights, in a riskless principal capacity, at such place and upon such terms (including public or private sale) as it may deem practicable. The Company shall assist the Depositary to the extent necessary to determine such legality and practicability. The Depositary shall, upon such sale, convert and distribute proceeds of such sale (net of applicable (a) fees and charges of, and expenses incurred by, the Depositary and (b) taxes) upon the terms set forth in Section 4.1.
(c) Lapse of Rights. If the Depositary is unable to make any rights available to Holders upon the terms described in Section 4.4(a) or to arrange for the sale of the rights upon the terms described in Section 4.4(b), the Depositary shall allow such rights to lapse.
The Depositary shall not be liable for (i) any failure to accurately determine whether it may be lawful or practicable to make such rights available to Holders in general or any Holders in particular, (ii) any foreign exchange exposure or loss incurred in connection with such sale, or exercise, or (iii) the content of any materials forwarded to the Holders on behalf of the Company in connection with the rights distribution.
Notwithstanding anything to the contrary in this Section 4.4, if registration (under the Securities Act or any other applicable law) of the rights or the securities to which any rights relate may be required in order for the Company to offer such rights or such securities to Holders and to sell the securities represented by such rights, the Depositary will not distribute such rights to the Holders (i) unless and until a registration statement under the Securities Act (or other applicable law) covering such offering is in effect or (ii) unless the Company furnishes the Depositary opinion(s) of counsel for the Company in the United States and counsel to the Company in any other applicable country in which rights would be distributed, in each case satisfactory to the Depositary, to the effect that the offering and sale of such securities to Holders and Beneficial Owners are exempt from, or do not require registration under, the provisions of the Securities Act or any other applicable laws.
In the event that the Company, the Depositary or the Custodian shall be required to withhold and does withhold from any distribution of Deposited Property (including rights) an amount on account of taxes or other governmental charges, the amount distributed to the Holders of ADSs shall be reduced accordingly. In the event that the Depositary determines that any distribution of Deposited Property (including Shares and rights to subscribe therefor) is subject to any tax or other governmental charges which the Depositary is obligated to withhold, the Depositary may dispose of all or a portion of such Deposited Property (including Shares and rights to subscribe therefor) in such amounts and in such manner, including by public or private sale, as the Depositary deems necessary and practicable to pay any such taxes or charges.
There can be no assurance that Holders generally, or any Holder in particular, will be given the opportunity to receive or exercise rights on the same terms and conditions as the holders of Shares or be able to exercise such rights. Nothing herein shall obligate the Company to file any registration statement in respect of any rights or Shares or other securities to be acquired upon the exercise of such rights.
Section 4.5 Distributions Other Than Cash, Shares or Rights to Purchase Shares.
(a) Whenever the Company intends to distribute to the holders of Deposited Securities property other than cash, Shares or rights to purchase additional Shares, the Company shall give timely notice thereof to the Depositary and shall indicate whether or not it wishes such distribution to be made to Holders of ADSs. Upon receipt of a notice indicating that the Company wishes such distribution to be made to Holders of ADSs, the Depositary shall consult with the Company, and the Company shall assist the Depositary, to determine whether such distribution to Holders is lawful and reasonably practicable. The Depositary shall not make such distribution unless (i) the Company shall have requested the Depositary to make such distribution to Holders, (ii) the Depositary shall have received satisfactory documentation within the terms of Section 5.7, and (iii) the Depositary shall have determined that such distribution is reasonably practicable.
(b) Upon receipt of satisfactory documentation and the request of the Company to distribute property to Holders of ADSs and after making the requisite determinations set forth in (a) above, the Depositary shall distribute the property so received to the Holders of record, as of the ADS Record Date, in proportion to the number of ADSs held by them respectively and in such manner as the Depositary may deem practicable for accomplishing such distribution (i) upon receipt of payment or net of the applicable fees and charges of, and expenses incurred by, the Depositary, and (ii) net of any applicable taxes required to be withheld. The Depositary may dispose of all or a portion of the property so distributed and deposited, in such amounts and in such manner (including public or private sale) as the Depositary may deem practicable or necessary to satisfy any taxes (including applicable interest and penalties) or other governmental charges applicable to the distribution.
(c) If (i) the Company does not request the Depositary to make such distribution to Holders or requests the Depositary not to make such distribution to Holders, (ii) the Depositary does not receive satisfactory documentation within the terms of Section 5.7, or (iii) the Depositary determines that all or a portion of such distribution is not reasonably practicable, the Depositary shall sell or cause such property to be sold in a public or private sale, at such place or places and upon such terms as it may deem practicable and shall (i) cause the proceeds of such sale, if any, to be converted into Dollars and (ii) distribute the proceeds of such conversion received by the Depositary (net of applicable (a) fees and charges of, and expenses incurred by, the Depositary and (b) taxes) to the Holders as of the ADS Record Date upon the terms of Section 4.1. If the Depositary is unable to sell such property, the Depositary may dispose of such property for the account of the Holders in any way it deems reasonably practicable under the circumstances.
(d) Neither the Depositary nor the Company shall be liable for (i) any failure to accurately determine whether it is lawful or practicable to make the property described in this Section 4.5 available to Holders in general or any Holders in particular, nor (ii) any loss incurred in connection with the sale or disposal of such property.
Section 4.6 Distributions with Respect to Deposited Securities in Bearer Form. Subject to the terms of this Article IV, distributions in respect of Deposited Securities that are held by the Depositary or the Custodian in bearer form shall be made to the Depositary for the account of the respective Holders of ADS(s) with respect to which any such distribution is made upon due presentation by the Depositary or the Custodian to the Company of any relevant coupons, talons, or certificates. The Company shall promptly notify the Depositary of such distributions. The Depositary or the Custodian shall promptly present such coupons, talons or certificates, as the case may be, in connection with any such distribution.
Section 4.7 Redemption. If the Company intends to exercise any right of redemption in respect of any of the Deposited Securities, the Company shall give notice thereof to the Depositary at least sixty (60) days (or such other number of days as mutually agreed to in writing by the Depositary and the Company) prior to the intended date of redemption which notice shall set forth the particulars of the proposed redemption. Upon timely receipt of (i) such notice and (ii) satisfactory documentation given by the Company to the Depositary within the terms of Section 5.7, and only if after consultation between the Depositary and the Company, the Depositary shall have determined that such proposed redemption is practicable, the Depositary shall provide to each Holder a notice setting forth the intended exercise by the Company of the redemption rights and any other particulars set forth in the Company’s notice to the Depositary. The Depositary shall instruct the Custodian to present to the Company the Deposited Securities in respect of which redemption rights are being exercised against payment of the applicable redemption price. Upon receipt of confirmation from the Custodian that the redemption has taken place and that funds representing the redemption price have been received, the Depositary shall convert, transfer, and distribute the proceeds (net of applicable (a) fees and charges of, and the expenses incurred by, the Depositary, and (b) taxes), retire ADSs and cancel ADRs, if applicable, upon delivery of such ADSs by Holders thereof and the terms set forth in Sections 4.1 and 6.2. If less than all outstanding Deposited Securities are redeemed, the ADSs to be retired will be selected by lot or on a pro rata basis, as may be determined by the Depositary after consultation with the Company. The redemption price per ADS shall be the dollar equivalent of the per share amount received by the Depositary (adjusted to reflect the ADS(s)-to-Share(s) ratio) upon the redemption of the Deposited Securities represented by ADSs (subject to the terms of Section 4.8 and the applicable fees and charges of, and expenses incurred by, the Depositary, and taxes) multiplied by the number of Deposited Securities represented by each ADS redeemed.
Notwithstanding anything contained in the Deposit Agreement to the contrary, in the event the Company fails to give the Depositary timely notice of the proposed redemption provided for in this Section 4.7, the Depositary agrees to use commercially reasonable efforts to perform the actions contemplated in this Section 4.7, and the Company, the Holders and the Beneficial Owners acknowledge that the Depositary shall have no liability for the Depositary’s failure to perform the actions contemplated in this Section 4.7 where such notice has not been so timely given, other than its failure to use commercially reasonable efforts, as provided herein.
Section 4.8 Conversion of Foreign Currency. Whenever the Depositary or the Custodian shall receive Foreign Currency, by way of dividends or other distributions or the net proceeds from the sale of Deposited Property, which in the judgment of the Depositary can at such time be converted on a practicable basis, by sale or in any other manner that it may determine in accordance with applicable law, into Dollars transferable to the United States and distributable to the Holders entitled thereto, the Depositary shall convert or cause to be converted, by sale or in any other manner that it may reasonably determine, such Foreign Currency into Dollars, and shall distribute such Dollars (net of the fees and charges set forth in the Fee Schedule attached hereto as Exhibit B, and applicable taxes withheld) in accordance with the terms of the applicable sections of the Deposit Agreement. The Depositary and/or its agent (which may be a division, branch or Affiliate of the Depositary) may act as principal for any conversion of Foreign Currency. If the Depositary shall have distributed warrants or other instruments that entitle the holders thereof to such Dollars, the Depositary shall distribute such Dollars to the holders of such warrants and/or instruments upon surrender thereof for cancellation, in either case without liability for interest thereon. Such distribution may be made upon an averaged or other practicable basis without regard to any distinctions among Holders on account of any application of exchange restrictions or otherwise.
If such conversion or distribution generally or with regard to a particular Holder can be effected only with the approval or license of any government or agency thereof, the Depositary shall have authority to file such application for approval or license, if any, as it may deem desirable. In no event, however, shall the Depositary be obligated to make such a filing.
If at any time the Depositary shall determine that in its judgment the conversion of any Foreign Currency and the transfer and distribution of proceeds of such conversion received by the Depositary is not practicable or lawful, or if any approval or license of any governmental authority or agency thereof that is required for such conversion, transfer and distribution is denied or, in the opinion of the Depositary, not obtainable at a reasonable cost or within a reasonable period, the Depositary may, in its reasonable discretion, (i) make such conversion and distribution in Dollars to the Holders for whom such conversion, transfer and distribution is lawful and practicable, (ii) distribute the Foreign Currency (or an appropriate document evidencing the right to receive such Foreign Currency) to Holders for whom this is lawful and practicable, or (iii) hold (or cause the Custodian to hold) such Foreign Currency (without liability for interest thereon) for the respective accounts of the Holders entitled to receive the same.
Section 4.9 Fixing of ADS Record Date. Whenever (a) the Depositary shall receive notice of the fixing of a record date by the Company for the determination of holders of Deposited Securities entitled to receive any distribution (whether in cash, Shares, rights, or other distribution), (b) for any reason the Depositary causes a change in the number of Shares that are represented by each ADS, (c) the Depositary shall receive notice of any meeting of, or solicitation of consents or proxies of, holders of Shares or other Deposited Securities, or (d) the Depositary shall find it necessary or convenient in connection with the giving of any notice, solicitation of any consent or any other matter, the Depositary shall fix the record date (the “ADS Record Date”) for the determination of the Holders of ADS(s) who shall be entitled to receive such distribution, to give instructions for the exercise of voting rights at any such meeting, to give or withhold such consent, to receive such notice or solicitation or to otherwise take action, or to exercise the rights of Holders with respect to such changed number of Shares represented by each ADS. The Depositary shall make reasonable efforts to establish the ADS Record Date as closely as practicable to the applicable record date for the Deposited Securities (if any) set by the Company in England and Wales and shall not announce the establishment of any ADS Record Date prior to the relevant corporate action having been made public by the Company (if such corporate action affects the Deposited Securities). Subject to applicable law and the provisions of Section 4.1 through 4.8 and to the other terms and conditions of the Deposit Agreement, only the Holders of ADSs at the close of business in New York on such ADS Record Date shall be entitled to receive such distribution, to give such voting instructions, to receive such notice or solicitation, or otherwise take action.
Section 4.10 Voting of Deposited Securities. As soon as practicable after receipt of notice of any meeting at which the holders of Deposited Securities are entitled to vote, or of solicitation of consents or proxies from holders of Deposited Securities, the Depositary shall fix the ADS Record Date in respect of such meeting or solicitation of consent or proxy in accordance with Section 4.9. The Depositary shall, if requested by the Company in writing in a timely manner (the Depositary having no obligation to take any further action if the request shall not have been received by the Depositary at least thirty (30) days prior to the date of such vote or meeting), at the Company’s expense and provided no U.S. legal prohibitions exist, distribute to Holders as of the ADS Record Date: (a) such notice of meeting or solicitation of consent or proxy, (b) a statement that the Holders at the close of business on the ADS Record Date will be entitled, subject to any applicable law, the provisions of the Deposit Agreement, the Articles of Association of the Company and the provisions of or governing the Deposited Securities (which provisions, if any, shall be summarized in pertinent part by the Company), to instruct the Depositary as to the exercise of the voting rights, if any, pertaining to the Deposited Securities represented by such Holder’s ADSs, and (c) a brief statement as to the manner in which such voting instructions may be given to the Depositary or in which voting instructions may be deemed to have been given in accordance with this Section 4.10.
Notwithstanding anything contained in the Deposit Agreement or any ADR, with the Company’s prior written consent, the Depositary may, to the extent not prohibited by law or regulations, or by the requirements of any stock exchange on which the ADSs may be listed, in lieu of distribution of the materials provided to the Depositary in connection with any meeting of, or solicitation of consents or proxies from, holders of Deposited Securities, distribute to the Holders a notice that provides Holders with, or otherwise publicizes to Holders, instructions on how to retrieve such materials or receive such materials upon request (e.g., by reference to a website containing the materials for retrieval or a contact for requesting copies of the materials).
The Depositary has been advised by the Company that under the Articles of Association of the Company as in effect on the date of the Deposit Agreement, voting at any meeting of shareholders of the Company is by poll.
Voting instructions may be given only in respect of a number of ADSs representing an integral number of Deposited Securities. Upon the timely receipt from a Holder of ADSs as of the ADS Record Date of voting instructions in the manner specified by the Depositary, the Depositary shall endeavor, insofar as practicable and permitted under any applicable law, the provisions of the Deposit Agreement, the Articles of Association of the Company and the provisions of the Deposited Securities, to vote, or cause the Custodian to vote, the Deposited Securities (in person or by proxy) represented by such Holder’s ADSs in accordance with the voting instructions received from the Holder of the ADSs. If the Depositary does not receive voting instructions from a Holder as of the ADS Record Date on or before the date established by the Depositary for such purpose, such Holder shall be deemed, and the Depositary shall deem such Holder, to have instructed the Depositary to give a discretionary proxy to a person designated by the Company to vote the Deposited Securities; provided, however, that no such discretionary proxy shall be given by the Depositary with respect to any matter to be voted upon as to which the Company informs the Depositary that (i) the Company does not wish such proxy to be given, (ii) substantial opposition exists, or (iii) the rights of holders of Deposited Securities may be adversely affected.
Deposited Securities represented by ADSs for which no timely voting instructions are received by the Depositary from the Holder shall not be voted (except as otherwise contemplated herein). Neither the Depositary nor the Custodian shall under any circumstances exercise any discretion as to voting and neither the Depositary nor the Custodian shall vote, attempt to exercise the right to vote, or in any way make use of, for purposes of establishing a quorum or otherwise, the Deposited Securities represented by ADSs, except pursuant to and in accordance with the voting instructions timely received from Holders or as otherwise contemplated herein. If the Depositary timely receives voting instructions from a Holder which fail to specify the manner in which the Depositary is to vote the Deposited Securities represented by such Holder’s ADSs, the Depositary will deem such Holder (unless otherwise specified in the notice distributed to Holders) to have instructed the Depositary to vote in favor of the items set forth in such voting instructions.
Notwithstanding anything else contained herein, the Depositary shall, if so requested in writing by the Company, represent all Deposited Securities (whether or not voting instructions have been received in respect of such Deposited Securities from Holders as of the ADS Record Date) for the sole purpose of establishing quorum at a meeting of shareholders.
Notwithstanding anything else contained in the Deposit Agreement or any ADR, the Depositary shall not have any obligation to take any action with respect to any meeting, or solicitation of consents or proxies, of holders of Deposited Securities if the taking of such action would violate the laws of the United States or England and Wales. The Company agrees to take any and all actions reasonably necessary and as permitted by the laws of England and Wales to enable Holders and Beneficial Owners to exercise the voting rights accruing to the Deposited Securities and to deliver to the Depositary an opinion of U.S. counsel addressing any actions requested to be taken if so reasonably requested by the Depositary.
There can be no assurance that Holders generally or any Holder in particular will receive the notice described above with sufficient time to enable the Holder to return voting instructions to the Depositary in a timely manner.
Section 4.11 Changes Affecting Deposited Securities. Upon any change in nominal value, sub-division, cancellation, consolidation or any other reclassification of Deposited Securities, or upon any recapitalization, reorganization, merger, scheme of arrangement, consolidation or sale of assets affecting the Company or to which it is a party, any property which shall be received by the Depositary or the Custodian in exchange for, or in conversion of, or replacement of, or otherwise in respect of, such Deposited Securities shall, to the extent permitted by law, be treated as new Deposited Property under the Deposit Agreement, and the ADSs shall, subject to the provisions of the Deposit Agreement, any ADR(s) evidencing such ADSs and applicable law, represent the right to receive such additional or replacement Deposited Property. In giving effect to such change, sub-division, cancellation, consolidation or other reclassification of Deposited Securities, recapitalization, reorganization, merger, scheme of arrangement, consolidation or sale of assets, the Depositary may, with the Company’s approval, and shall, if the Company shall so request, subject to the terms of the Deposit Agreement (including, without limitation, (a) the applicable fees and charges of, and expenses incurred by, the Depositary, and (b) applicable taxes) and receipt of an opinion of counsel to the Company satisfactory to the Depositary that such actions are not in violation of any applicable laws or regulations, (i) issue and deliver additional ADSs as in the case of a stock dividend on the Shares, (ii) amend the Deposit Agreement and the applicable ADRs, (iii) amend the applicable Registration Statement(s) on Form F-6 as filed with the Commission in respect of the ADSs, (iv) call for the surrender of outstanding ADRs to be exchanged for new ADRs, and (v) take such other actions as are appropriate to reflect the transaction with respect to the ADSs. The Company agrees to, jointly with the Depositary, amend the Registration Statement on Form F-6 as filed with the Commission to permit the issuance of such new form of ADRs. Notwithstanding the foregoing, in the event that any Deposited Property so received may not be lawfully distributed to some or all Holders, the Depositary may, with the Company’s approval, and shall, if the Company requests, subject to receipt of an opinion of Company’s counsel satisfactory to the Depositary that such action is not in violation of any applicable laws or regulations, sell such Deposited Property at public or private sale, at such place or places and upon such terms as it may deem proper and may allocate the net proceeds of such sales (net of (a) fees and charges of, and expenses incurred by, the Depositary and (b) taxes) for the account of the Holders otherwise entitled to such Deposited Property upon an averaged or other practicable basis without regard to any distinctions among such Holders and distribute the net proceeds so allocated to the extent practicable as in the case of a distribution received in cash pursuant to Section 4.1. The Depositary shall not be responsible for (i) any failure to determine that it may be lawful or practicable to make such Deposited Property available to Holders in general or to any Holder in particular, (ii) any foreign exchange exposure or loss incurred in connection with such sale, or (iii) any liability to the purchaser of such Deposited Property.
Section 4.12 Available Information.
The Company is subject to the periodic reporting requirements of the Exchange Act and, accordingly, is required to file or furnish certain reports with the Commission. These reports can be retrieved from the Commission's website (www.sec.gov) and can be inspected and copied at the public reference facilities maintained by the Commission located (as of the date of the Deposit Agreement) at 100 F Street, N.E., Washington D.C. 20549.
Section 4.13 Reports. The Depositary shall make available for inspection by Holders at its Principal Office, as promptly as practicable after receipt thereof, any reports and communications, including any proxy soliciting materials, received from the Company which are both (a) received by the Depositary, the Custodian, or the nominee of either of them as the holder of the Deposited Property and (b) made generally available to the holders of such Deposited Property by the Company. The Depositary shall also provide or make available to Holders copies of such reports when furnished by the Company pursuant to Section 5.6.
Section 4.14 List of Holders. Promptly upon written request by the Company, the Depositary shall furnish to it a list, as of a recent date, of the names, addresses and holdings of ADSs of all Holders.
Section 4.15 Taxation. The Depositary will, and will instruct the Custodian to, forward to the Company or its agents such information from its records as the Company may reasonably request to enable the Company or its agents to file the necessary tax reports with governmental authorities or agencies. The Depositary, the Custodian or the Company and its agents may file such reports as are necessary to reduce or eliminate applicable taxes on dividends and on other distributions in respect of Deposited Property under applicable tax treaties or laws for the Holders and Beneficial Owners. In accordance with instructions from the Company and to the extent practicable, the Depositary or the Custodian will take reasonable administrative actions to obtain tax refunds, reduced withholding of tax at source on dividends and other benefits under applicable tax treaties or laws with respect to dividends and other distributions on the Deposited Property. As a condition to receiving such benefits, Holders and Beneficial Owners of ADSs may be required from time to time, and in a timely manner, to file such proof of taxpayer status, residence and beneficial ownership (as applicable), to execute such certificates and to make such representations and warranties, or to provide any other information or documents, as the Depositary or the Custodian may deem necessary or proper to fulfill the Depositary’s or the Custodian’s obligations under applicable law. The Depositary and the Company shall have no obligation or liability to any person if any Holder or Beneficial Owner fails to provide such information or if such information does not reach the relevant tax authorities in time for any Holder or Beneficial Owner to obtain the benefits of any tax treatment. The Holders and Beneficial Owners shall indemnify the Depositary, the Company, the Custodian and any of their respective directors, employees, agents and Affiliates against, and hold each of them harmless from, any claims by any governmental authority with respect to taxes, additions to tax, penalties or interest arising out of any refund of taxes, reduced rate of withholding at source or other tax benefit obtained.
If the Company (or any of its agents) withholds from any distribution any amount on account of taxes or governmental charges, or pays any other tax in respect of such distribution (e.g., stamp duty tax, capital gains or other similar tax), the Company shall use commercially reasonable efforts to (or shall cause such agent to) remit within a reasonable time to the Depositary information about such taxes or governmental charges withheld or paid, and, if so reasonably requested, the tax receipt (or other proof of payment to the applicable governmental authority) therefor, in each case, in a form reasonably satisfactory to the Depositary. The Depositary shall, to the extent required by U.S. law, report to Holders any taxes withheld by it or the Custodian, and, if such information is provided to it by the Company, any taxes withheld by the Company. The Depositary and the Custodian shall not be required to provide the Holders with any evidence of the remittance by the Company (or its agents) of any taxes withheld, or of the payment of taxes by the Company, except to the extent the evidence is provided by the Company to the Depositary or the Custodian, as applicable. Neither the Depositary nor the Custodian shall be liable for the failure by any Holder or Beneficial Owner to obtain the benefits of credits on the basis of non-U.S. tax paid against such Holder’s or Beneficial Owner’s income tax liability.
The Depositary is under no obligation to provide the Holders and Beneficial Owners with any information about the tax status of the Company except to the extent that the Company provides information to the Depositary for distribution to the Holders and Beneficial Owners, and the Depositary reasonably agrees to distribute to the Holders and Beneficial Owners. The Depositary shall not incur any liability for any tax consequences that may be incurred by Holders and Beneficial Owners on account of their ownership of the ADSs, including without limitation, tax consequences resulting from the Company (or any of its subsidiaries) being treated as a “Passive Foreign Investment Company” (in each case as defined in the U.S. Internal Revenue Code and the regulations issued thereunder) or otherwise.
ARTICLE V
THE DEPOSITARY, THE CUSTODIAN AND THE COMPANY
Section 5.1 Maintenance of Office and Transfer Books by the Registrar. Until termination of the Deposit Agreement in accordance with its terms, the Registrar shall maintain in the Borough of Manhattan, the City of New York, an office and facilities for the issuance and delivery of ADSs, the acceptance for surrender of ADS(s) for the purpose of withdrawal of Deposited Securities, the registration of issuances, cancellations, transfers, combinations and split-ups of ADS(s) and, if applicable, to countersign ADRs evidencing the ADSs so issued, transferred, combined or split-up, in each case in accordance with the provisions of the Deposit Agreement.
The Registrar shall keep books for the registration of ADSs which at all reasonable times shall be open for inspection by the Company and by the Holders of such ADSs, provided that such inspection shall not be, to the Registrar’s knowledge, for the purpose of communicating with Holders of such ADSs in the interest of a business or object other than the business of the Company or other than a matter related to the Deposit Agreement or the ADSs.
The Registrar may close the transfer books with respect to the ADSs, at any time or from time to time, when deemed necessary or advisable by it in good faith in connection with the performance of its duties hereunder, or at the reasonable written request of the Company subject, in all cases, to Section 7.8(a).
If any ADSs are listed on one or more stock exchanges or automated quotation systems in the United States, the Depositary shall act as Registrar or, with written notice given as promptly as practicable to the Company, appoint a Registrar or one or more co-registrars for registration of issuances, cancellations, transfers, combinations and split-ups of ADSs and, if applicable, to countersign ADRs evidencing the ADSs so issued, transferred, combined or split-up, in accordance with any requirements of such exchanges or systems. Such Registrar or co-registrars may be removed and a substitute or substitutes appointed by the Depositary, upon written notice given as promptly as practicable to the Company.
Section 5.2 Exoneration. Notwithstanding anything contained in the Deposit Agreement or any ADR, neither the Depositary nor the Company shall be obligated to do or perform any act or thing which is inconsistent with the provisions of the Deposit Agreement or incur any liability (to the extent not limited by Section 7.8(b)) (i) if the Depositary, the Custodian, the Company or their respective agents shall be prevented or forbidden from, hindered or delayed in, doing or performing any act or thing required or contemplated by the terms of the Deposit Agreement, by reason of any provision of any present or future law or regulation of the United States, England and Wales or any other country, or of any other governmental authority or regulatory authority or stock exchange, or on account of potential criminal or civil penalties or restraint, or by reason of any provision, present or future, of the Articles of Association of the Company or any provision of or governing any Deposited Securities, or by reason of any act of God or other event or circumstance beyond its control (including, without limitation, fire, flood, earthquake, tornado, hurricane, tsunami, explosion, or other natural disaster, nationalization, expropriation, currency restriction, work stoppage, strikes, civil unrest, act of war (whether declared or not) or terrorism, revolution, rebellion, embargo, computer failure, failure of public infrastructure (including communication or utility failure), failure of common carriers, nuclear, cyber or biochemical incident, any pandemic, epidemic or other prevalent disease or illness with an actual or probable threat to human life, any quarantine order or travel restriction imposed by a governmental authority or other competent public health authority, or the failure or unavailability of the United States Federal Reserve Bank (or other central banking system) or DTC (or other clearing system)), (ii) by reason of any exercise of, or failure to exercise, any discretion provided for in the Deposit Agreement or in the Articles of Association of the Company or provisions of or governing Deposited Securities, (iii) for any action or inaction in reliance upon the advice of or information from legal counsel, accountants, any person presenting Shares for deposit, any Holder, any Beneficial Owner or authorized representative thereof, or any other person believed by it in good faith to be competent to give such advice or information, (iv) for the inability by a Holder or Beneficial Owner to benefit from any distribution, offering, right or other benefit which is made available to holders of Deposited Securities but is not, under the terms of the Deposit Agreement, made available to Holders of ADSs, (v) for any action or inaction of any clearing or settlement system (and any participant thereof) for the Deposited Property or the ADSs, or (vi) for any consequential or punitive damages (including lost profits) for any breach of the terms of the Deposit Agreement.
The Depositary, its controlling persons, its agents, any Custodian and the Company, its controlling persons and its agents may rely and shall be protected in acting upon any written notice, request or other document believed by it to be genuine and to have been signed or presented by the proper party or parties.
Section 5.3 Standard of Care. The Company and the Depositary assume no obligation and shall not be subject to any liability under the Deposit Agreement or any ADRs to any Holder(s) or Beneficial Owner(s), except that the Company and the Depositary agree to perform their respective obligations specifically set forth in the Deposit Agreement or the applicable ADRs without negligence or bad faith.
Without limitation of the foregoing, neither the Depositary, nor the Company, nor any of their respective controlling persons, or agents, shall be under any obligation to appear in, prosecute or defend any action, suit or other proceeding in respect of any Deposited Property or in respect of the ADSs, which in its opinion may involve it in expense or liability, unless indemnity satisfactory to it against all expense (including fees and disbursements of counsel) and liability be furnished as often as may be required (and no Custodian shall be under any obligation whatsoever with respect to such proceedings, the responsibility of the Custodian being solely to the Depositary).
The Depositary and its agents shall not be liable for any failure to carry out any instructions to vote any of the Deposited Securities, or for the manner in which any vote is cast or the effect of any vote, provided that any such action or omission is in good faith and without negligence and in accordance with the terms of the Deposit Agreement. The Depositary shall not incur any liability for any failure to accurately determine that any distribution or action may be lawful or reasonably practicable, for the content of any information submitted to it by the Company for distribution to the Holders or for any inaccuracy of any translation thereof, for any investment risk associated with acquiring an interest in the Deposited Property, for the validity or worth of the Deposited Property, for the value of any Deposited Property or any distribution thereon, for any interest on Deposited Property, for any tax consequences that may result from the ownership of ADSs, Shares or other Deposited Property, for the credit-worthiness of any third party, for allowing any rights to lapse upon the terms of the Deposit Agreement, for the failure or timeliness of any notice from the Company, or for any action of or failure to act by, or any information provided or not provided by, DTC or any DTC Participant.
The Depositary shall not be liable for any acts or omissions made by a successor depositary in connection with any matter arising wholly prior to the appointment of the Depositary or after the removal or resignation of the Depositary, provided that in connection with the issue out of which such potential liability arises, the Depositary performed its obligations without negligence or bad faith while it acted as Depositary for the Company.
Section 5.4 Resignation and Removal of the Depositary; Appointment of Successor Depositary. The Depositary may at any time resign as Depositary hereunder by written notice of resignation delivered to the Company, such resignation to be effective on the earlier of (i) the 90th day after delivery thereof to the Company (whereupon the Depositary shall be entitled to take the actions contemplated in Section 6.2), or (ii) the appointment by the Company of a successor depositary and its acceptance of such appointment as hereinafter provided.
The Depositary may at any time be removed by the Company by written notice of such removal, which removal shall be effective on the later of (i) the 90th day after delivery thereof to the Depositary (whereupon the Depositary shall be entitled to take the actions contemplated in Section 6.2), or (ii) upon the appointment by the Company of a successor depositary and its acceptance of such appointment as hereinafter provided.
In case at any time the Depositary acting hereunder shall resign or be removed, the Company shall use its best efforts to appoint a successor depositary, which shall be a bank or trust company having an office in the Borough of Manhattan, the City of New York. Every successor depositary shall be required by the Company to execute and deliver to its predecessor and to the Company an instrument in writing accepting its appointment hereunder, and thereupon such successor depositary, without any further act or deed (except as required by applicable law), shall become fully vested with all the rights, powers, duties and obligations of its predecessor (other than as contemplated in Sections 5.8 and 5.9). The predecessor depositary, upon payment of all sums due it and on the written request of the Company, shall, (i) execute and deliver an instrument transferring to such successor all rights and powers of such predecessor hereunder (other than as contemplated in Sections 5.8 and 5.9), (ii) duly assign, transfer and deliver all of the Depositary’s right, title and interest to the Deposited Property to such successor, and (iii) deliver to such successor a list of the Holders of all outstanding ADSs and such other information relating to ADSs and Holders thereof as the successor may reasonably request. Any such successor depositary shall promptly provide notice of its appointment to such Holders.
Any entity into or with which the Depositary may be merged or consolidated shall be the successor of the Depositary without the execution or filing of any document or any further act.
Section 5.5 The Custodian. The Depositary has initially appointed Citibank, N.A. (London) as Custodian for the purpose of the Deposit Agreement. The Custodian or its successors in acting hereunder shall be authorized to act as custodian in England and Wales and shall be subject at all times and in all respects to the direction of the Depositary for the Deposited Property for which the Custodian acts as custodian and shall be responsible solely to it. If any Custodian resigns or is discharged from its duties hereunder with respect to any Deposited Property and no other Custodian has previously been appointed hereunder, the Depositary shall promptly appoint a substitute custodian. The Depositary shall require such resigning or discharged Custodian to Deliver, or cause the Delivery of, the Deposited Property held by it, together with all such records maintained by it as Custodian with respect to such Deposited Property as the Depositary may request, to the Custodian designated by the Depositary. Whenever the Depositary determines, in its discretion, that it is appropriate to do so, it may appoint an additional custodian with respect to any Deposited Property, or discharge the Custodian with respect to any Deposited Property and appoint a substitute custodian, which shall thereafter be Custodian hereunder with respect to the Deposited Property. Immediately upon any such change, the Depositary shall give notice thereof in writing to all Holders of ADSs, each other Custodian and the Company.
Citibank may at any time act as Custodian of the Deposited Property pursuant to the Deposit Agreement, in which case any reference to Custodian shall mean Citibank solely in its capacity as Custodian pursuant to the Deposit Agreement. Notwithstanding anything contained in the Deposit Agreement or any ADR to the contrary, the Depositary shall not be obligated to give notice to the Company, any Holders of ADSs or any other Custodian of its acting as Custodian pursuant to the Deposit Agreement.
Upon the appointment of any successor depositary, any Custodian then acting hereunder shall, unless otherwise instructed by the Depositary, continue to be the Custodian of the Deposited Property without any further act or writing, and shall be subject to the direction of the successor depositary. The successor depositary so appointed shall, nevertheless, on the written request of any Custodian, execute and deliver to such Custodian all such instruments as may be proper to give to such Custodian full and complete power and authority to act on the direction of such successor depositary.
Section 5.6 Notices and Reports. On or before the first date on which the Company gives notice, by publication or otherwise, of any meeting of holders of Shares or other Deposited Securities, or of any adjourned meeting of such holders, or of the taking of any action by such holders other than at a meeting, or of the taking of any action in respect of any cash or other distributions or the offering of any rights in respect of Deposited Securities, the Company shall transmit to the Depositary and the Custodian a copy of the notice thereof in the English language but otherwise in the form given or to be given to holders of Shares or other Deposited Securities. The Company shall also furnish to the Custodian and the Depositary a summary, in English, of any applicable provisions or proposed provisions of the Articles of Association of the Company that may be relevant or pertain to such notice of meeting or be the subject of a vote thereat.
The Depositary shall arrange, at the request of the Company and at the Company’s expense, to provide copies thereof to all Holders or make such notices, reports and other communications available to all Holders on a basis similar to that for holders of Shares or other Deposited Securities or on such other basis as the Company may advise the Depositary or as may be required by any applicable law, regulation or stock exchange requirement. The Company has delivered to the Depositary and the Custodian a copy of the Company’s Articles of Association along with the provisions of or governing the Shares and any other Deposited Securities issued by the Company in connection with such Shares, and promptly upon any amendment thereto or change therein, the Company shall deliver to the Depositary and the Custodian a copy of such amendment thereto or change therein. The Depositary may rely upon such copy for all purposes of the Deposit Agreement.
The Depositary will, at the expense of the Company, make available a copy of any such notices, reports or communications issued by the Company and delivered to the Depositary for inspection by the Holders of the ADSs at the Depositary’s Principal Office, at the office of the Custodian and at any other designated transfer office.
Section 5.7 Issuance of Additional Shares, ADSs etc. The Company agrees that in the event it or any of its Affiliates proposes (i) an issuance, sale or distribution of additional Shares, (ii) an offering of rights to subscribe for Shares or other Deposited Securities, (iii) an issuance or assumption of securities convertible into or exchangeable for Shares, (iv) an issuance of rights to subscribe for securities convertible into or exchangeable for Shares, (v) an elective dividend of cash or Shares, (vi) a redemption of Deposited Securities, (vii) a meeting of holders of Deposited Securities, or solicitation of consents or proxies, relating to any reclassification of securities, merger, scheme of arrangement or consolidation or transfer of assets, (viii) any assumption, reclassification, recapitalization, reorganization, merger, scheme of arrangement, consolidation or sale of assets which affects the Deposited Securities, or (ix) a distribution of securities other than Shares, it will obtain U.S. legal advice and take all steps necessary to ensure that the application of the proposed transaction to Holders and Beneficial Owners does not violate the registration provisions of the Securities Act, or any other applicable laws (including, without limitation, the Investment Company Act of 1940, as amended, the Exchange Act and the securities laws of the states of the U.S.). In support of the foregoing, the Company will furnish to the Depositary (a) a written opinion of U.S. counsel (reasonably satisfactory to the Depositary) stating whether such transaction (1) requires a registration statement under the Securities Act to be in effect or (2) is exempt from the registration requirements of the Securities Act and (b) an opinion of English counsel stating that (1) making the transaction available to Holders and Beneficial Owners does not violate the laws or regulations of England and Wales and (2) all requisite regulatory consents and approvals have been obtained in England and Wales. If the filing of a registration statement is required, the Depositary shall not have any obligation to proceed with the transaction unless it shall have received evidence reasonably satisfactory to it that such registration statement has been declared effective. If, being advised by counsel, the Company determines that a transaction is required to be registered under the Securities Act, the Company will either (i) register such transaction to the extent necessary, (ii) alter the terms of the transaction to avoid the registration requirements of the Securities Act or (iii) direct the Depositary to take specific measures, in each case as contemplated in the Deposit Agreement, to prevent such transaction from violating the registration requirements of the Securities Act. The Company agrees with the Depositary that neither the Company nor any of its Affiliates will at any time (i) deposit any Shares or other Deposited Securities, either upon original issuance or upon a sale of Shares or other Deposited Securities previously issued and reacquired by the Company or by any such Affiliate, or (ii) issue additional Shares, rights to subscribe for such Shares, securities convertible into or exchangeable for Shares or rights to subscribe for such securities or distribute securities other than Shares, unless such transaction and the securities issuable in such transaction do not violate the registration provisions of the Securities Act, or any other applicable laws (including, without limitation, the Investment Company Act of 1940, as amended, the Exchange Act and the securities laws of the states of the U.S.).
Notwithstanding anything else contained in the Deposit Agreement, nothing in the Deposit Agreement shall be deemed to obligate the Company to file any registration statement in respect of any proposed transaction.
Section 5.8 Indemnification. The Depositary agrees to indemnify the Company and its directors, officers, employees, agents and Affiliates against, and hold each of them harmless from, any direct loss, liability, tax, charge or expense of any kind whatsoever (including, but not limited to, the reasonable fees and expenses of counsel) which may arise out of acts performed or omitted by the Depositary under the terms hereof due to the negligence or bad faith of the Depositary.
The Company agrees to indemnify the Depositary, the Custodian and any of their respective directors, officers, employees, agents and Affiliates against, and hold each of them harmless from, any direct loss, liability, tax, charge or expense of any kind whatsoever (including, but not limited to, the reasonable fees and expenses of counsel) that may arise (a) out of, or in connection with, any offer, issuance, sale, resale, transfer, deposit or withdrawal of ADRs, ADSs, the Shares, or other Deposited Securities, as the case may be, to the extent that it is not unlawful for the Company to indemnify such person at such time under the applicable laws of England and Wales, (b) out of, or as a result of, any offering documents in respect thereof or (c) out of acts performed or omitted, including, but not limited to, any delivery by the Depositary on behalf of the Company of information regarding the Company, in connection with the Deposit Agreement, any ancillary or supplemental agreement entered into between the Company and the Depositary, the ADRs, the ADSs, the Shares, or any Deposited Property, in any such case (i) by the Depositary, the Custodian or any of their respective directors, officers, employees, agents and Affiliates, except to the extent such loss, liability, tax, charge or expense is due to the negligence or bad faith of any of them, or (ii) by the Company or any of its directors, officers, employees, agents and Affiliates. Notwithstanding the foregoing, the Company shall not indemnify the Depositary or the Custodian (for so long as the Custodian is a branch of Citibank, N.A.) against any fees, charges or expenses payable by third party Holders or Beneficial Owners under this Deposit Agreement including, for the avoidance of doubt, those fees and charges set out in the Fee Schedule attached hereto as Exhibit B.
The obligations set forth in this Section shall survive the termination of the Deposit Agreement and the succession or substitution of any party hereto.
Any person seeking indemnification hereunder (an “indemnified person”) shall notify the person from whom it is seeking indemnification (the “indemnifying person”) of the commencement of any indemnifiable action or claim promptly after such indemnified person becomes aware of such commencement (provided that the failure to make such notification shall not affect such indemnified person’s rights to seek indemnification except to the extent the indemnifying person is materially prejudiced by such failure) and shall consult in good faith with the indemnifying person as to the conduct of the defense of such action or claim that may give rise to an indemnity hereunder, which defense shall be reasonable in the circumstances. No indemnified person shall compromise or settle any action or claim that may give rise to an indemnity hereunder without the consent of the indemnifying person, which consent shall not be unreasonably withheld.
Section 5.9 ADS Fees and Charges. The Company, the Holders, the Beneficial Owners, persons depositing Shares or withdrawing Deposited Securities in connection with the issuance and cancellation of ADSs, and persons receiving ADSs upon issuance or whose ADSs are being cancelled shall be required to pay the Depositary’s fees and related charges identified as payable by them respectively in the Fee Schedule attached hereto as Exhibit B. All ADS fees and charges so payable may be deducted from distributions or must be remitted to the Depositary, or its designee, and may, at any time and from time to time, be changed by agreement between the Depositary and the Company, but, in the case of ADS fees and charges payable by Holders and Beneficial Owners, only in the manner contemplated in Section 6.1. The Depositary shall provide, without charge, a copy of its latest ADS fee schedule to anyone upon request.
ADS fees and charges payable for (i) the issuance of ADSs and (ii) the cancellation of ADSs will be payable by the person for whom the ADSs are so issued by the Depositary (in the case of ADS issuances) and by the person for whom ADSs are being cancelled (in the case of ADS cancellations). In the case of ADSs issued by the Depositary into DTC or presented to the Depositary via DTC, the ADS issuance and cancellation fees and charges will be payable by the DTC Participant(s) receiving the ADSs from the Depositary or the DTC Participant(s) holding the ADSs being cancelled, as the case may be, on behalf of the Beneficial Owner(s) and will be charged by the DTC Participant(s) to the account(s) of the applicable Beneficial Owner(s) in accordance with the procedures and practices of the DTC Participant(s) as in effect at the time. ADS fees and charges in respect of distributions and the ADS service fee are payable by Holders as of the applicable ADS Record Date established by the Depositary. In the case of distributions of cash, the amount of the applicable ADS fees and charges is deducted from the funds being distributed. In the case of (i) distributions other than cash and (ii) the ADS service fee, the applicable Holders as of the ADS Record Date established by the Depositary will be invoiced for the amount of the ADS fees and charges and such ADS fees may be deducted from distributions made to Holders. For ADSs held through DTC, the ADS fees and charges for distributions other than cash and the ADS service fee may be deducted from distributions made through DTC, and may be charged to the DTC Participants in accordance with the procedures and practices prescribed by DTC from time to time and the DTC Participants in turn charge the amount of such ADS fees and charges to the Beneficial Owners for whom they hold ADSs. In the case of (i) registration of ADS transfers, the ADS transfer fee will be payable by the ADS Holder whose ADSs are being transferred or by the person to whom the ADSs are transferred, and (ii) conversion of ADSs of one series for ADSs of another series, the ADS conversion fee will be payable by the Holder whose ADSs are converted or by the person to whom the converted ADSs are delivered.
The Depositary may reimburse the Company for certain expenses incurred by the Company in respect of the ADR program established pursuant to the Deposit Agreement, by making available a portion of the ADS fees charged in respect of the ADR program or otherwise, upon such terms and conditions as the Company and the Depositary agree from time to time. The Company shall pay to the Depositary such fees and charges, and reimburse the Depositary for such out-of-pocket expenses, as the Depositary and the Company may agree from time to time. Responsibility for payment of such fees, charges and reimbursements may from time to time be changed by agreement between the Company and the Depositary. Unless otherwise agreed, the Depositary shall present its statement for such fees, charges and reimbursements to the Company once every three months. The charges and expenses of the Custodian are for the sole account of the Depositary.
The obligations of Holders and Beneficial Owners to pay ADS fees and charges shall survive the termination of the Deposit Agreement. As to any Depositary, upon the resignation or removal of such Depositary as described in Section 5.4, the right to collect ADS fees and charges shall extend for those ADS fees and charges incurred prior to the effectiveness of such resignation or removal.
Section 5.10 Restricted Securities Owners. The Company agrees to advise in writing each of the persons or entities who, to the knowledge of the Company, holds Restricted Securities that such Restricted Securities are ineligible for deposit hereunder (except under the circumstances contemplated in Section 2.14) and, to the extent practicable, shall require each of such persons to represent in writing that such person will not deposit Restricted Securities hereunder (except under the circumstances contemplated in Section 2.14).
ARTICLE VI
AMENDMENT AND TERMINATION
Section 6.1 Amendment/Supplement. Subject to the terms and conditions of this Section 6.1 and applicable law, the ADRs outstanding at any time, the provisions of the Deposit Agreement and the form of ADR attached hereto and to be issued under the terms hereof may at any time and from time to time be amended or supplemented by written agreement between the Company and the Depositary in any respect which they may deem necessary or desirable without the prior written consent of the Holders or Beneficial Owners. Any amendment or supplement which shall impose or increase any fees or charges (other than charges in connection with foreign exchange control regulations, and taxes and other governmental charges, delivery and other such expenses), or which shall otherwise materially prejudice any substantial existing right of Holders or Beneficial Owners, shall not, however, become effective as to outstanding ADSs until the expiration of thirty (30) days after notice of such amendment or supplement shall have been given to the Holders of outstanding ADSs. Notice of any amendment to the Deposit Agreement or any ADR shall not need to describe in detail the specific amendments effectuated thereby, and failure to describe the specific amendments in any such notice shall not render such notice invalid, provided, however, that, in each such case, the notice given to the Holders identifies a means for Holders and Beneficial Owners to retrieve or receive the text of such amendment (e.g., upon retrieval from the Commission’s, the Depositary’s or the Company’s website or upon request from the Depositary). The parties hereto agree that any amendments or supplements which (i) are reasonably necessary (as agreed by the Company and the Depositary) in order for (a) the ADSs to be registered on Form F-6 under the Securities Act or (b) the ADSs to be settled solely in electronic book-entry form and (ii) do not in either such case impose or increase any fees or charges to be borne by Holders, shall be deemed not to materially prejudice any substantial existing rights of Holders or Beneficial Owners. Every Holder and Beneficial Owner at the time any amendment or supplement so becomes effective shall be deemed, by continuing to hold such ADSs, to consent and agree to such amendment or supplement and to be bound by the Deposit Agreement and the ADR, if applicable, as amended or supplemented thereby. In no event shall any amendment or supplement impair the right of the Holder to surrender such ADS and receive therefor the Deposited Securities represented thereby, except in order to comply with mandatory provisions of applicable law. Notwithstanding the foregoing, if any governmental body should adopt new laws, rules or regulations which would require an amendment of, or supplement to, the Deposit Agreement to ensure compliance therewith, the Company and the Depositary may amend or supplement the Deposit Agreement and any ADRs at any time in accordance with such changed laws, rules or regulations. Such amendment or supplement to the Deposit Agreement and any ADRs in such circumstances may become effective before a notice of such amendment or supplement is given to Holders or within any other period of time as required for compliance with such laws, rules or regulations.
Section 6.2 Termination. The Depositary shall, at any time at the written direction of the Company, terminate the Deposit Agreement by distributing notice of such termination to the Holders of all ADSs then outstanding at least thirty (30) days prior to the date fixed in such notice for such termination. If (i) ninety (90) days shall have expired after the Depositary shall have delivered to the Company a written notice of its election to resign, or (ii) ninety (90) days shall have expired after the Company shall have delivered to the Depositary a written notice of the removal of the Depositary, and, in either case, a successor depositary shall not have been appointed and accepted its appointment as provided in Section 5.4 of the Deposit Agreement, the Depositary may terminate the Deposit Agreement by distributing notice of such termination to the Holders of all ADSs then outstanding at least thirty (30) days prior to the date fixed in such notice for such termination. The date so fixed for termination of the Deposit Agreement in any termination notice so distributed by the Depositary to the Holders of ADSs is referred to as the “Termination Date”. Until the Termination Date, the Depositary shall continue to perform all of its obligations under the Deposit Agreement, and the Holders and Beneficial Owners will be entitled to all of their rights under the Deposit Agreement.
If any ADSs shall remain outstanding after the Termination Date, the Registrar and the Depositary shall not, after the Termination Date, have any obligation to perform any further acts under the Deposit Agreement, except that the Depositary shall, subject, in each case, to the terms and conditions of the Deposit Agreement, continue to (i) collect dividends and other distributions pertaining to Deposited Securities, (ii) sell Deposited Property received in respect of Deposited Securities, (iii) deliver Deposited Securities, together with any dividends or other distributions received with respect thereto and the net proceeds of the sale of any other Deposited Property, in exchange for ADSs surrendered to the Depositary (after deducting, or charging, as the case may be, in each case, the fees and charges of, and expenses incurred by, the Depositary, and all applicable taxes or governmental charges for the account of the Holders and Beneficial Owners, in each case upon the terms set forth in Section 5.9 of the Deposit Agreement), and (iv) take such actions as may be required under applicable law in connection with its role as Depositary under the Deposit Agreement.
At any time after the Termination Date, the Depositary may sell the Deposited Property then held under the Deposit Agreement and shall after such sale hold un-invested the net proceeds of such sale, together with any other cash then held by it under the Deposit Agreement, in an un-segregated account and without liability for interest, for the pro rata benefit of the Holders whose ADSs have not theretofore been surrendered. After making such sale, the Depositary shall be discharged from all obligations under the Deposit Agreement except (i) to account for such net proceeds and other cash (after deducting, or charging, as the case may be, in each case, the fees and charges of, and expenses incurred by, the Depositary, and all applicable taxes or governmental charges for the account of the Holders and Beneficial Owners, in each case upon the terms set forth in Section 5.9 of the Deposit Agreement), and (ii) as may be required at law in connection with the termination of the Deposit Agreement. After the Termination Date, the Company shall be discharged from all obligations under the Deposit Agreement, except for its obligations to the Depositary under Sections 5.8, 5.9 and 7.6 of the Deposit Agreement. The obligations under the terms of the Deposit Agreement of Holders and Beneficial Owners of ADSs outstanding as of the Termination Date shall survive the Termination Date and shall be discharged only when the applicable ADSs are presented by their Holders to the Depositary for cancellation under the terms of the Deposit Agreement (except as specifically provided in the Deposit Agreement).
Notwithstanding anything contained in the Deposit Agreement or any ADR, in connection with the termination of the Deposit Agreement, the Depositary may, independently and without the need for any action by the Company, make available to Holders of ADSs a means to withdraw the Deposited Securities represented by their ADSs and to direct the deposit of such Deposited Securities into an unsponsored American depositary shares program established by the Depositary, upon such terms and conditions as the Depositary may deem reasonably appropriate, subject however, in each case, to satisfaction of the applicable registration requirements by the unsponsored American depositary shares program under the Securities Act, and to receipt by the Depositary of payment of the applicable fees and charges of, and reimbursement of the applicable expenses incurred by, the Depositary.
ARTICLE VII
MISCELLANEOUS
Section 7.1 Counterparts. The Deposit Agreement may be executed in any number of counterparts, each of which shall be deemed an original and all of such counterparts together shall constitute one and the same agreement. Copies of the Deposit Agreement shall be maintained with the Depositary and shall be open to inspection by any Holder during business hours.
Section 7.2 No Third-Party Beneficiaries/Acknowledgments. The Deposit Agreement is for the exclusive benefit of the parties hereto (and their successors) and shall not be deemed to give any legal or equitable right, remedy or claim whatsoever to any other person, except to the extent specifically set forth in the Deposit Agreement. Nothing in the Deposit Agreement shall be deemed to give rise to a partnership or joint venture among the parties nor establish a fiduciary or similar relationship among the parties. The parties hereto acknowledge and agree that (i) Citibank and its Affiliates may at any time have multiple banking relationships with the Company, the Holders, the Beneficial Owners, and their respective Affiliates, (ii) Citibank and its Affiliates may own and deal in any class of securities of the Company and its Affiliates and in ADSs, and may be engaged at any time in transactions in which parties adverse to the Company, the Holders, the Beneficial Owners or their respective Affiliates may have interests, (iii) the Depositary and its Affiliates may from time to time have in their possession non-public information about the Company, the Holders, the Beneficial Owners, and their respective Affiliates, (iv) nothing contained in the Deposit Agreement shall (a) preclude Citibank or any of its Affiliates from engaging in such transactions or establishing or maintaining such relationships, or (b) obligate Citibank or any of its Affiliates to disclose such information, transactions or relationships, or to account for any profit made or payment received in such transactions or relationships, (v) the Depositary shall not be deemed to have knowledge of any information any other division of Citibank or any of its Affiliates may have about the Company, the Holders, the Beneficial Owners, or any of their respective Affiliates, and (vi) the Company, the Depositary, the Custodian and their respective agents and controlling persons may be subject to the laws and regulations of jurisdictions other than the United States and England and Wales, and the authority of courts and regulatory authorities of such other jurisdictions, and, consequently, the requirements and the limitations of such other laws and regulations, and the decisions and orders of such other courts and regulatory authorities, may affect the rights and obligations of the parties to the Deposit Agreement.
Section 7.3 Severability. In case any one or more of the provisions contained in the Deposit Agreement or in the ADRs should be or become invalid, illegal or unenforceable in any respect, the validity, legality and enforceability of the remaining provisions contained herein or therein shall in no way be affected, prejudiced or disturbed thereby.
The Depositary may execute transactions contemplated herein (e.g., foreign currency conversions, and sales of Deposited Property) through one or more divisions of Citibank or through one or more Citibank Affiliates, and any such entity may act as principal for its own account and not as agent, advisor, broker or fiduciary on behalf of any other person and may earn and retain revenue from such transactions, including, without, without limitation, transaction spreads, commissions, etc. The Depositary does not guarantee or represent that the price or rate obtained in any such transaction, or the method for obtaining such price or rate, will be the most favorable that could be obtained at that time.
Section 7.4 Holders and Beneficial Owners as Parties; Binding Effect. The Holders and Beneficial Owners from time to time of ADSs issued hereunder shall be parties to the Deposit Agreement and shall be bound by all of the terms and conditions hereof and of any ADR evidencing their ADSs by acceptance thereof or any beneficial interest therein.
Section 7.5 Notices. Any and all notices to be given to the Company shall be deemed to have been duly given if personally delivered or sent by mail, air courier or cable, telex or facsimile transmission, confirmed by letter personally delivered or sent by mail or air courier, addressed to Immunocore Holdings plc, 92 Park Drive, Milton Park, Abingdon, Oxfordshire, United Kingdom OX14 4RY, Attention: Lily Hepworth, General Counsel and Company Secretary, with a copy (which shall not constitute notice) to Cooley LLP, 55 Hudson Yards, New York, New York 10001, Attention: Divakar Gupta, or to any other address which the Company may specify in writing to the Depositary.
Any and all notices to be given to the Depositary shall be deemed to have been duly given if personally delivered or sent by mail, air courier or cable, telex or facsimile transmission, confirmed by letter personally delivered or sent by mail or air courier, addressed to Citibank, N.A., 388 Greenwich Street, New York, New York 10013, U.S.A., Attention: Depositary Receipts Department, or to any other address which the Depositary may specify in writing to the Company.
Any and all notices to be given to any Holder shall be deemed to have been duly given (a) if personally delivered or sent by mail or cable, telex or facsimile transmission, confirmed by letter, addressed to such Holder at the address of such Holder as it appears on the books of the Depositary or, if such Holder shall have filed with the Depositary a request that notices intended for such Holder be mailed to some other address, at the address specified in such request, or (b) if a Holder shall have designated such means of notification as an acceptable means of notification under the terms of the Deposit Agreement, by means of electronic messaging addressed for delivery to the e-mail address designated by the Holder for such purpose. Notice to Holders shall be deemed to be notice to Beneficial Owners for all purposes of the Deposit Agreement. Failure to notify a Holder or any defect in the notification to a Holder shall not affect the sufficiency of notification to other Holders or to the Beneficial Owners of ADSs held by such other Holders. Any notices given to DTC under the terms of the Deposit Agreement shall (unless otherwise specified by the Depositary) constitute notice to the DTC Participants who hold the ADSs in their DTC accounts and to the Beneficial Owners of such ADSs.
Delivery of a notice sent by electronic mail, mail, air courier or cable, telex or facsimile transmission shall be deemed to be effective at the time when a duly addressed letter containing the same (or a confirmation thereof in the case of a cable, telex or facsimile transmission) is deposited, postage prepaid, in a post-office letter box or delivered to an air courier service, without regard for the actual receipt or time of actual receipt thereof by a Holder. The Depositary or the Company may, however, act upon any cable, telex or facsimile transmission received by it from any Holder, the Custodian, the Depositary, or the Company, notwithstanding that such cable, telex or facsimile transmission shall not be subsequently confirmed by letter.
Delivery of a notice by means of electronic messaging shall be deemed to be effective at the time of the initiation of the transmission by the sender (as shown on the sender’s records), notwithstanding that the intended recipient retrieves the message at a later date, fails to retrieve such message, or fails to receive such notice on account of its failure to maintain the designated e-mail address, its failure to designate a substitute e-mail address or for any other reason.
Section 7.6 Governing Law and Jurisdiction. The Deposit Agreement, the ADRs and the ADSs shall be interpreted in accordance with, and all rights hereunder and thereunder and provisions hereof and thereof shall be governed by, the laws of the State of New York applicable to contracts made and to be wholly performed in that State. Notwithstanding anything contained in the Deposit Agreement to the contrary, any ADR or any present or future provisions of the laws of the State of New York, the rights of holders of Shares and of any other Deposited Securities and the obligations and duties of the Company in respect of the holders of Shares and other Deposited Securities, as such, shall be governed by the laws of England and Wales (or, if applicable, such other laws as may govern the Deposited Securities).
Except as set forth in the following paragraph of this Section 7.6, the Company and the Depositary agree that the federal or state courts in the City of New York shall have jurisdiction to hear and determine any suit, action or proceeding and to settle any dispute between them that may arise out of or in connection with the Deposit Agreement and, for such purposes, each irrevocably submits to the non-exclusive jurisdiction of such courts. The Company hereby irrevocably designates, appoints and empowers Immunocore, LLC (the “Agent”) now at Six Tower Bridge, Suite 200, 181 Washington Street, Conshohocken, Pennsylvania 19428, United States as its authorized agent to receive and accept for and on its behalf, and on behalf of its properties, assets and revenues, service by mail of any and all legal process, summons, notices and documents that may be served in any suit, action or proceeding brought against the Company in any federal or state court as described in the preceding sentence or in the next paragraph of this Section 7.6. If for any reason the Agent shall cease to be available to act as such, the Company agrees to designate a new agent in New York on the terms and for the purposes of this Section 7.6 reasonably satisfactory to the Depositary. The Company further hereby irrevocably consents and agrees to the service of any and all legal process, summons, notices and documents in any suit, action or proceeding against the Company, by service by mail of a copy thereof upon the Agent (whether or not the appointment of such Agent shall for any reason prove to be ineffective or such Agent shall fail to accept or acknowledge such service), with a copy mailed to the Company by registered or certified air mail, postage prepaid, to its address provided in Section 7.5. The Company agrees that the failure of the Agent to give any notice of such service to it shall not impair or affect in any way the validity of such service or any judgment rendered in any action or proceeding based thereon.
Notwithstanding the foregoing, the Depositary and the Company unconditionally agree that in the event that a Holder or Beneficial Owner brings a suit, action or proceeding against (a) the Company, (b) the Depositary in its capacity as Depositary under the Deposit Agreement, or (c) against both the Company and the Depositary, in any such case, in any state or federal court of the United States, and the Depositary or the Company have any claim, for indemnification or otherwise, against each other arising out of the subject matter of such suit, action or proceeding, then the Company and the Depositary may pursue such claim against each other in the state or federal court in the United States in which such suit, action, or proceeding is pending and, for such purposes, the Company and the Depositary irrevocably submit to the non-exclusive jurisdiction of such courts. The Company agrees that service of process upon the Agent in the manner set forth in the preceding paragraph shall be effective service upon it for any suit, action or proceeding brought against it as described in this paragraph.
The Company irrevocably and unconditionally waives, to the fullest extent permitted by law, any objection that it may now or hereafter have to the laying of venue of any actions, suits or proceedings brought in any court as provided in this Section 7.6, and hereby further irrevocably and unconditionally waives and agrees not to plead or claim in any such court that any such action, suit or proceeding brought in any such court has been brought in an inconvenient forum.
The Company irrevocably and unconditionally waives, to the fullest extent permitted by law, and agrees not to plead or claim, any right of immunity from legal action, suit or proceeding, from setoff or counterclaim, from the jurisdiction of any court, from service of process, from attachment upon or prior to judgment, from attachment in aid of execution or judgment, from execution of judgment, or from any other legal process or proceeding for the giving of any relief or for the enforcement of any judgment, and consents to such relief and enforcement against it, its assets and its revenues in any jurisdiction, in each case with respect to any matter arising out of, or in connection with, the Deposit Agreement, any ADR or the Deposited Property.
EACH OF THE PARTIES TO THE DEPOSIT AGREEMENT (INCLUDING, WITHOUT LIMITATION, EACH HOLDER AND BENEFICIAL OWNER) IRREVOCABLY WAIVES, TO THE FULLEST EXTENT PERMITTED BY APPLICABLE LAW, ANY AND ALL RIGHT TO TRIAL BY JURY IN ANY LEGAL PROCEEDING AGAINST THE COMPANY AND/OR THE DEPOSITARY ARISING OUT OF, OR RELATING TO, THE DEPOSIT AGREEMENT, ANY ADR AND ANY TRANSACTIONS CONTEMPLATED THEREIN (WHETHER BASED ON CONTRACT, TORT, COMMON LAW OR OTHERWISE).
The provisions of this Section 7.6 shall survive any termination of the Deposit Agreement, in whole or in part.
Section 7.7 Assignment. Subject to the provisions of Section 5.4, the Deposit Agreement may not be assigned by either the Company or the Depositary.
Section 7.8 Compliance with, and No Disclaimer under, U.S. Securities Laws.
(a) Notwithstanding anything in the Deposit Agreement to the contrary, the withdrawal or delivery of Deposited Securities will not be suspended by the Company or the Depositary except as would be permitted by Instruction I.A.(1) of the General Instructions to Form F-6 Registration Statement, as amended from time to time, under the Securities Act.
(b) Each of the parties to the Deposit Agreement (including, without limitation, each Holder and Beneficial Owner) acknowledges and agrees that no provision of the Deposit Agreement or any ADR shall, or shall be deemed to, disclaim any liability under the Securities Act or the Exchange Act, in each case to the extent established under applicable U.S. laws.
Section 7.9 English Law References. Any summary of the laws and regulations of England and Wales and of the terms of the Company’s Articles of Association set forth in the Deposit Agreement have been provided by the Company solely for the convenience of Holders, Beneficial Owners and the Depositary. While such summaries are believed by the Company to be accurate as of the date of the Deposit Agreement, (i) they are summaries and as such may not include all aspects of the materials summarized applicable to a Holder or Beneficial Owner, and (ii) these laws and regulations and the Company’s Articles of Association may change after the date of the Deposit Agreement. Neither the Depositary nor the Company has any obligation under the terms of the Deposit Agreement to update any such summaries.
Section 7.10 Titles and References.
(a) Deposit Agreement. All references in the Deposit Agreement to exhibits, articles, sections, subsections, and other subdivisions refer to the exhibits, articles, sections, subsections and other subdivisions of the Deposit Agreement unless expressly provided otherwise. The words “the Deposit Agreement”, “herein”, “hereof”, “hereby”, “hereunder”, and words of similar import refer to the Deposit Agreement as a whole as in effect at the relevant time between the Company, the Depositary and the Holders and Beneficial Owners of ADSs and not to any particular subdivision unless expressly so limited. Pronouns in masculine, feminine and neuter gender shall be construed to include any other gender, and words in the singular form shall be construed to include the plural and vice versa unless the context otherwise requires. Titles to sections of the Deposit Agreement are included for convenience only and shall be disregarded in construing the language contained in the Deposit Agreement. References to “applicable laws and regulations” shall refer to laws and regulations applicable to ADRs, ADSs or Deposited Property as in effect at the relevant time of determination, unless otherwise required by law or regulation.
(b) ADRs. All references in any ADR(s) to paragraphs, exhibits, articles, sections, subsections, and other subdivisions refer to the paragraphs, exhibits, articles, sections, subsections and other subdivisions of the ADR(s) in question unless expressly provided otherwise. The words “the Receipt”, “the ADR”, “herein”, “hereof”, “hereby”, “hereunder”, and words of similar import used in any ADR refer to the ADR as a whole and as in effect at the relevant time, and not to any particular subdivision unless expressly so limited. Pronouns in masculine, feminine and neuter gender in any ADR shall be construed to include any other gender, and words in the singular form shall be construed to include the plural and vice versa unless the context otherwise requires. Titles to paragraphs of any ADR are included for convenience only and shall be disregarded in construing the language contained in the ADR. References to “applicable laws and regulations” shall refer to laws and regulations applicable to the Company, the Depositary, the Custodian, their agents and controlling persons, the ADRs, the ADSs and the Deposited Property as in effect at the relevant time of determination, unless otherwise required by law or regulation.
[Signature Page Follows]
IN WITNESS WHEREOF, Immunocore Holdings PLC and CITIBANK, N.A. have duly executed the Deposit Agreement as of the day and year first above set forth and all Holders and Beneficial Owners shall become parties hereto upon acceptance by them of ADSs issued in accordance with the terms hereof, or upon acquisition of any beneficial interest therein.
|
Immunocore Holdings PLC
|
|||
| By: |
/s/ Brian Di Donato |
||
| |
Name: |
Brian Di Donato |
|
| |
Title: |
Chief Financial Officer |
|
|
CITIBANK, N.A
|
|||
| By: |
/s/ Leslie DeLuca |
||
| |
Name: |
Leslie DeLuca |
|
| |
Title: |
Attorney-in-Fact |
|
[DEPOSIT AGREEMENT]
EXHIBIT A
[FORM OF ADR]
| Number | CUSIP NUMBER: _______ |
| _____________ | |
| American Depositary Shares (each American Depositary Share representing the right to receive one (1) fully paid ordinary share) |
AMERICAN DEPOSITARY RECEIPT
for
AMERICAN DEPOSITARY SHARES
representing
DEPOSITED ORDINARY SHARES
of
Immunocore Holdings PLC
(Incorporated under the laws of England and Wales)
CITIBANK, N.A., a national banking association organized and existing under the laws of the United States of America, as depositary (the “Depositary”), hereby certifies that _____________is the owner of ______________ American Depositary Shares (hereinafter “ADS”) representing deposited ordinary shares, including evidence of rights to receive such ordinary shares (the “Shares”), of Immunocore Holdings PLC, a public limited company incorporated under the laws of England and Wales (the “Company”). As of the date of issuance of this ADR, each ADS represents the right to receive one (1) Share deposited under the Deposit Agreement (as hereinafter defined) with the Custodian, which at the date of issuance of this ADR is Citibank, N.A. (London) (the “Custodian”). The ADS(s)-to-Share(s) ratio is subject to amendment as provided in Articles IV and VI of the Deposit Agreement. The Depositary’s Principal Office is located at 388 Greenwich Street, New York, New York 10013, U.S.A.
(1) The Deposit Agreement. This American Depositary Receipt is one of an issue of American Depositary Receipts (“ADRs”), all issued and to be issued upon the terms and conditions set forth in the Deposit Agreement, dated as of February 9, 2021 (as amended and supplemented from time to time, the “Deposit Agreement”), by and among the Company, the Depositary, and all Holders and Beneficial Owners from time to time of ADSs issued thereunder. The Deposit Agreement sets forth the rights and obligations of Holders and Beneficial Owners of ADSs and the rights and duties of the Depositary in respect of the Shares deposited thereunder and any and all other Deposited Property (as defined in the Deposit Agreement) from time to time received and held on deposit in respect of the ADSs. Copies of the Deposit Agreement are on file at the Principal Office of the Depositary and with the Custodian. Each Holder and each Beneficial Owner, upon acceptance of any ADSs (or any interest therein) issued in accordance with the terms and conditions of the Deposit Agreement, shall be deemed for all purposes to (a) be a party to and bound by the terms of the Deposit Agreement and the applicable ADR(s), and (b) appoint the Depositary its attorney-in-fact, with full power to delegate, to act on its behalf and to take any and all actions contemplated in the Deposit Agreement and the applicable ADR(s), to adopt any and all procedures necessary to comply with applicable law and to take such action as the Depositary in its sole discretion may deem necessary or appropriate to carry out the purposes of the Deposit Agreement and the applicable ADR(s), the taking of such actions to be the conclusive determinant of the necessity and appropriateness thereof. The manner in which a Beneficial Owner holds ADSs (e.g., in a brokerage account vs. as registered holder) may affect the rights and obligations of, the manner in which, and the extent to which, services are made available to, Beneficial Owners pursuant to the terms of the Deposit Agreement.
The statements made on the face and reverse of this ADR are summaries of certain provisions of the Deposit Agreement and the Articles of Association of the Company (as in effect on the date of the signing of the Deposit Agreement) and are qualified by and subject to the detailed provisions of the Deposit Agreement and the Articles of Association of the Company, to which reference is hereby made.
All capitalized terms not defined herein shall have the meanings ascribed thereto in the Deposit Agreement.
The Depositary makes no representation or warranty as to the validity or worth of the Deposited Property. The Depositary has made arrangements for the acceptance of the ADSs into DTC. Each Beneficial Owner of ADSs held through DTC must rely on the procedures of DTC and the DTC Participants to exercise and be entitled to any rights attributable to such ADSs. The Depositary may issue Uncertificated ADSs subject, however, to the terms and conditions of Section 2.13 of the Deposit Agreement.
(2) Surrender of ADSs and Withdrawal of Deposited Securities. The Holder of this ADR (and of the ADSs evidenced hereby) shall be entitled to Delivery (at the Custodian’s designated office) of the Deposited Securities at the time represented by the ADSs evidenced hereby upon satisfaction of each of the following conditions: (i) the Holder (or a duly-authorized attorney of the Holder) has duly Delivered ADSs to the Depositary at its Principal Office the ADSs evidenced hereby (and, if applicable, this ADR evidencing such ADSs) for the purpose of withdrawal of the Deposited Securities represented thereby, (ii) if applicable and so required by the Depositary, this ADR Delivered to the Depositary for such purpose has been properly endorsed in blank or is accompanied by proper instruments of transfer in blank (including signature guarantees in accordance with standard securities industry practice), (iii) if so required by the Depositary, the Holder of the ADSs has executed and delivered to the Depositary a written order directing the Depositary to cause the Deposited Securities being withdrawn to be Delivered to or upon the written order of the person(s) designated in such order, and (iv) all applicable fees and charges of, and expenses incurred by, the Depositary and all applicable taxes and governmental charges (as are set forth in Section 5.9 of, and Exhibit B to, the Deposit Agreement) have been paid, subject, however, in each case, to the terms and conditions of this ADR evidencing the surrendered ADSs, of the Deposit Agreement, of the Company’s Articles of Association and of any applicable laws and the rules of CREST, and to any provisions of or governing the Deposited Securities, in each case as in effect at the time thereof.
Upon satisfaction of each of the conditions specified above, the Depositary (i) shall cancel the ADSs Delivered to it (and, if applicable, this ADR(s) evidencing the ADSs so Delivered), (ii) shall direct the Registrar to record the cancellation of the ADSs so Delivered on the books maintained for such purpose, and (iii) shall direct the Custodian to Deliver, or cause the Delivery of, in each case, without unreasonable delay, the Deposited Securities represented by the ADSs so canceled together with any certificate or other document of title for the Deposited Securities, or evidence of the electronic transfer thereof (if available), as the case may be, to or upon the written order of the person(s) designated in the order delivered to the Depositary for such purpose, subject however, in each case, to the terms and conditions of the Deposit Agreement, of this ADR evidencing the ADS so canceled, of the Articles of Association of the Company, of any applicable laws and of the rules of CREST, and to the terms and conditions of or governing the Deposited Securities, in each case as in effect at the time thereof.
The Depositary shall not accept for surrender ADSs representing less than one (1) Share. In the case of Delivery to it of ADSs representing a number other than a whole number of Shares, the Depositary shall cause ownership of the appropriate whole number of Shares to be Delivered in accordance with the terms hereof, and shall, at the discretion of the Depositary, either (i) return to the person surrendering such ADSs the number of ADSs representing any remaining fractional Share, or (ii) sell or cause to be sold the fractional Share represented by the ADSs so surrendered and remit the proceeds of such sale (net of (a) applicable fees and charges of, and expenses incurred by, the Depositary and (b) applicable taxes required to be withheld as a result of such sale) to the person surrendering the ADSs.
Notwithstanding anything else contained in this ADR or the Deposit Agreement, the Depositary may make delivery at the Principal Office of the Depositary of Deposited Property consisting of (i) any cash dividends or cash distributions, or (ii) any proceeds from the sale of any non-cash distributions, which are at the time held by the Depositary in respect of the Deposited Securities represented by the ADSs surrendered for cancellation and withdrawal. At the request, risk and expense of any Holder so surrendering ADSs represented by this ADR, and for the account of such Holder, the Depositary shall direct the Custodian to forward (to the extent permitted by law) any Deposited Property (other than Deposited Securities) held by the Custodian in respect of such ADSs to the Depositary for delivery at the Principal Office of the Depositary. Such direction shall be given by letter or, at the request, risk and expense of such Holder, by cable, telex or facsimile transmission.
(3) Transfer, Combination and Split-up of ADRs. The Registrar shall register the transfer of this ADR (and of the ADSs represented hereby) on the books maintained for such purpose and the Depositary shall (x) cancel this ADR and execute new ADRs evidencing the same aggregate number of ADSs as those evidenced by this ADR canceled by the Depositary, (y) cause the Registrar to countersign such new ADRs, and (z) Deliver such new ADRs to or upon the order of the person entitled thereto, if each of the following conditions has been satisfied: (i) this ADR has been duly Delivered by the Holder (or by a duly authorized attorney of the Holder) to the Depositary at its Principal Office for the purpose of effecting a transfer thereof, (ii) this surrendered ADR has been properly endorsed or is accompanied by proper instruments of transfer (including signature guarantees in accordance with standard securities industry practice), (iii) this surrendered ADR has been duly stamped (if required by the laws of the State of New York or of the United States), and (iv) all applicable fees and charges of, and expenses incurred by, the Depositary and all applicable taxes and governmental charges (as are set forth in Section 5.9 of, and Exhibit B to, the Deposit Agreement) have been paid, subject, however, in each case, to the terms and conditions of this ADR, of the Deposit Agreement and of applicable law, in each case as in effect at the time thereof.
The Registrar shall register the split-up or combination of this ADR (and of the ADSs represented hereby) on the books maintained for such purpose and the Depositary shall (x) cancel this ADR and execute new ADRs for the number of ADSs requested, but in the aggregate not exceeding the number of ADSs evidenced by this ADR canceled by the Depositary, (y) cause the Registrar to countersign such new ADRs, and (z) Deliver such new ADRs to or upon the order of the Holder thereof, if each of the following conditions has been satisfied: (i) this ADR has been duly Delivered by the Holder (or by a duly authorized attorney of the Holder) to the Depositary at its Principal Office for the purpose of effecting a split-up or combination hereof, and (ii) all applicable fees and charges of, and expenses incurred by, the Depositary and all applicable taxes and governmental charges (as are set forth in Section 5.9 of, and Exhibit B to, the Deposit Agreement) have been paid, subject, however, in each case, to the terms and conditions of this ADR, of the Deposit Agreement and of applicable law, in each case as in effect at the time thereof.
(4) Pre-Conditions to Registration, Transfer, Etc. As a condition precedent to the execution and Delivery, the registration of issuance, transfer, split-up, combination or surrender, of any ADS, the delivery of any distribution thereon, or the withdrawal of any Deposited Property, the Depositary or the Custodian may require (i) payment from the depositor of Shares or presenter of ADSs or of this ADR of a sum sufficient to reimburse it for any tax or other governmental charge and any stock transfer or registration fee with respect thereto (including any such tax or charge and fee with respect to Shares being deposited or withdrawn) and payment of any applicable fees and charges of the Depositary as provided in Section 5.9 and Exhibit B to the Deposit Agreement and in this ADR, (ii) the production of proof reasonably satisfactory to it as to the identity and genuineness of any signature or any other matter contemplated by Section 3.1 of the Deposit Agreement, and (iii) compliance with (A) any laws or governmental regulations relating to the execution and Delivery of this ADR or ADSs or to the withdrawal of Deposited Securities and (B) such reasonable regulations as the Depositary and the Company may establish consistent with the provisions of this ADR, if applicable, the Deposit Agreement and applicable law.
The issuance of ADSs against deposits of Shares generally or against deposits of particular Shares may be suspended, or the deposit of particular Shares may be refused, or the registration of transfer of ADSs in particular instances may be refused, or the registration of transfer of ADSs generally may be suspended, during any period when the transfer books of the Company, the Depositary, a Registrar or the Share Registrar are closed or if any such action is deemed necessary or advisable by the Depositary or the Company, in good faith, at any time or from time to time because of any requirement of law or regulation, any government or governmental body or commission or any securities exchange on which the ADSs or Shares are listed, or under any provision of the Deposit Agreement or this ADR, if applicable, or under any provision of, or governing, the Deposited Securities, or because of a meeting of shareholders of the Company or for any other reason, subject, in all cases to Section 7.8 of the Deposit Agreement and paragraph (25) of this ADR. Notwithstanding any provision of the Deposit Agreement or this ADR to the contrary, Holders are entitled to surrender outstanding ADSs to withdraw the Deposited Securities associated therewith at any time subject only to (i) temporary delays caused by closing the transfer books of the Depositary or the Company or the deposit of Shares in connection with voting at a shareholders’ meeting or the payment of dividends, (ii) the payment of fees, taxes and similar charges, (iii) compliance with any U.S. or foreign laws or governmental regulations relating to the ADSs or to the withdrawal of the Deposited Securities, and (iv) other circumstances specifically contemplated by Instruction I.A.(l) of the General Instructions to Form F-6 (as such General Instructions may be amended from time to time).
(5) Compliance With Information Requests. Notwithstanding any other provision of the Deposit Agreement or this ADR, each Holder and Beneficial Owner of the ADSs represented hereby agrees to comply with requests from the Company pursuant to applicable law, the rules and requirements of any stock exchange on which the Shares or ADSs are, or will be, registered, traded or listed, and/or the Articles of Association of the Company, which are made to provide information, inter alia, as to the capacity in which such Holder or Beneficial Owner owns ADSs (and the Shares represented by such ADSs, as the case may be) and regarding the identity of any other person(s) interested in such ADSs (and the Shares represented by such ADSs, as the case may be) and the nature of such interest and various other matters, whether or not they are Holders and/or Beneficial Owners at the time of such request.
(6) Ownership Restrictions. Notwithstanding any other provision contained in this ADR or of the Deposit Agreement to the contrary, the Company may restrict transfers of the Shares where such transfer might result in ownership of Shares exceeding limits imposed by applicable law or the Articles of Association of the Company. The Company may also restrict, in such manner as it deems appropriate, transfers of the ADSs where such transfer may result in the total number of Shares represented by the ADSs owned by a single Holder or Beneficial Owner to exceed any such limits. The Company may, in its sole discretion but subject to applicable law, instruct the Depositary to take action with respect to the ownership interest of any Holder or Beneficial Owner in excess of the limits set forth in the preceding sentence, including but not limited to, the imposition of restrictions on the transfer of ADSs, the removal or limitation of voting rights or the mandatory sale or disposition on behalf of a Holder or Beneficial Owner of the Shares represented by the ADSs held by such Holder or Beneficial Owner in excess of such limitations, if and to the extent such disposition is permitted by applicable law and the Articles of Association of the Company. Nothing herein or in the Deposit Agreement shall be interpreted as obligating the Depositary or the Company to ensure compliance with the ownership restrictions described herein or in Section 3.5 of the Deposit Agreement.
Notwithstanding any provision of the Deposit Agreement or of this ADRs and without limiting the foregoing, by being a Holder or Beneficial Owner of an ADS, each such Holder or Beneficial Owner agrees to provide such information as the Company may request in a disclosure notice (a “Disclosure Notice”) given pursuant to the U.K. Companies Act 2006 (as amended from time to time and including any statutory modification or re-enactment thereof, the “Companies Act”) or the Articles of Association of the Company. By accepting or holding an ADS, each Holder and Beneficial Owner acknowledges that it understands that failure to comply with a Disclosure Notice may result in the imposition of sanctions against the holder of the Shares in respect of which the non-complying person is or was, or appears to be or has been, interested as provided in the Companies Act and the Articles of Association which currently include, as of the date of this ADR, the withdrawal of the voting rights of such Shares and (where the relevant Shares represent at least 0.25% in nominal value of the issued shares of their class (calculated exclusive of any shares held as treasury shares)) the imposition of restrictions on the rights to receive dividends on and to transfer such Shares.
The Company reserves the right to instruct Holders and Beneficial Owners to deliver their ADSs for cancellation and withdrawal of the Deposited Securities so as to permit the Company to deal directly with the Holder and Beneficial Owner thereof as a holder of Shares and Holders agree to comply with such instructions. The Depositary agrees to cooperate with the Company in its efforts to inform Holders and Beneficial Owners of the Company’s exercise of its rights under this paragraph and agrees to consult with, and provide reasonable assistance without risk, liability or expense on the part of the Depositary, to the Company on the manner or manners in which it may enforce such rights with respect to any Holder or Beneficial Owner.
(7) Reporting Obligations and Regulatory Approvals. Applicable laws and regulations may require holders and beneficial owners of Shares, including the Holders and Beneficial Owners of ADSs, to satisfy reporting requirements and obtain regulatory approvals in certain circumstances. Holders and Beneficial Owners of ADSs are solely responsible for determining and complying with such reporting requirements and obtaining such approvals. Each Holder and each Beneficial Owner hereby agrees to make such determination, file such reports, and obtain such approvals to the extent and in the form required by applicable laws and regulations as in effect from time to time. Neither the Depositary, the Custodian, the Company or any of their respective agents or Affiliates shall be required to take any actions whatsoever on behalf of Holders or Beneficial Owners to determine or satisfy such reporting requirements or obtain such regulatory approvals under applicable laws and regulations.
(8) Liability for Taxes and Other Charges. Any tax or other governmental charge payable by the Custodian or by the Depositary with respect to any Deposited Property, ADSs or this ADR shall be payable by the Holders and Beneficial Owners to the Depositary. The Company, the Custodian and/or the Depositary may withhold or deduct from any distributions made in respect of Deposited Property held on behalf of such Holder and/or Beneficial Owner, and may sell for the account of a Holder and/or Beneficial Owner any or all of such Deposited Property and apply such distributions and sale proceeds in payment of, any taxes (including applicable interest and penalties) or charges that are or may be payable by Holders or Beneficial Owners in respect of the ADSs, Deposited Property and this ADR, the Holder and the Beneficial Owner hereof remaining liable for any deficiency. The Custodian may refuse the deposit of Shares and the Depositary may refuse to issue ADSs, to deliver ADRs, register the transfer of ADSs, register the split-up or combination of ADRs and (subject to paragraph (25) of this ADR and Section 7.8 of the Deposit Agreement) the withdrawal of Deposited Property until payment in full of such tax, charge, penalty or interest is received. Every Holder and Beneficial Owner agrees to indemnify the Depositary, the Company, the Custodian, and any of their agents, officers, employees and Affiliates for, and to hold each of them harmless from, any claims with respect to taxes (including applicable interest and penalties thereon) arising from (i) any ADS held by such Holder and/or owned by such Beneficial Owner, (ii) the Deposited Property represented by the ADSs, and (iii) any transaction entered into by such Holder and/or Beneficial Owner in respect of the ADSs and/or the Deposited Property represented thereby. Notwithstanding anything to the contrary contained in the Deposit Agreement or any ADR, the obligations of Holders and Beneficial Owners under Section 3.2 of the Deposit Agreement shall survive any transfer of ADSs, any cancellation of ADSs and withdrawal of Deposited Securities, and the termination of the Deposit Agreement.
(9) Representations and Warranties on Deposit of Shares. Each person depositing Shares under the Deposit Agreement shall be deemed thereby to represent and warrant that (i) such Shares and the certificates therefor are duly authorized, validly issued, fully paid, non-assessable (i.e., not subject to call for payment of further capital) and legally obtained by such person, (ii) all preemptive (and similar) rights, if any, with respect to such Shares have been validly waived or exercised, (iii) the person making such deposit is duly authorized so to do, (iv) the Shares presented for deposit are free and clear of any lien, encumbrance, security interest, charge, mortgage or adverse claim, (v) the Shares presented for deposit are not, and the ADSs issuable upon such deposit will not be, Restricted Securities (except as contemplated in Section 2.14 of the Deposit Agreement), (vi) the Shares presented for deposit have not been stripped of any rights or entitlements, and (vii) the deposit of the Shares does not violate any applicable provisions of English law. Such representations and warranties shall survive the deposit and withdrawal of Shares, the issuance and cancellation of ADSs in respect thereof and the transfer of such ADSs. If any such representations or warranties are false in any way, the Company and the Depositary shall be authorized, at the cost and expense of the person depositing Shares, to take any and all actions necessary to correct the consequences thereof.
(10) Proofs, Certificates and Other Information. Any person presenting Shares for deposit, any Holder and any Beneficial Owner may be required, and every Holder and Beneficial Owner agrees, from time to time to provide to the Depositary and the Custodian such proof of citizenship or residence, taxpayer status, payment of all applicable taxes or other governmental charges, exchange control approval, legal or beneficial ownership of ADSs and Deposited Property, compliance with applicable laws, the terms of the Deposit Agreement or this ADR evidencing the ADSs and the provisions of, or governing, the Deposited Property, to execute such certifications and to make such representations and warranties, and to provide such other information and documentation (or, in the case of Shares in registered form presented for deposit, such information relating to the registration on the books of the Company or of the Share Registrar) as the Depositary or the Custodian may deem necessary or proper or as the Company may reasonably require by written request to the Depositary consistent with its obligations under the Deposit Agreement and this ADR. The Depositary and the Registrar, as applicable, may, and at the reasonable request of the Company, shall, to the extent practicable, withhold the execution or delivery or registration of transfer of any ADR or ADS or the distribution or sale of any dividend or distribution of rights or of the proceeds thereof or, to the extent not limited by paragraph (25) and Section 7.8 of the Deposit Agreement, the delivery of any Deposited Property until such proof or other information is filed or such certifications are executed, or such representations and warranties are made or such other documentation or information are provided, in each case to the Depositary’s, the Registrar’s and the Company’s satisfaction.
(11) ADS Fees and Charges. The following ADS fees are payable under the terms of the Deposit Agreement:
| (i) | ADS Issuance Fee: by any person for whom ADSs are issued (e.g., an issuance upon a deposit of Shares, upon a change in the ADS(s)-to-Share(s) ratio, or for any other reason), excluding issuances as a result of distributions described in paragraph (iv) below, a fee not in excess of U.S. $5.00 per 100 ADSs (or fraction thereof) issued under the terms of the Deposit Agreement; |
| (ii) | ADS Cancellation Fee: by any person for whom ADSs are being cancelled (e.g., a cancellation of ADSs for Delivery of deposited shares, upon a change in the ADS(s)-to-Share(s) ratio, or for any other reason), a fee not in excess of U.S. $5.00 per 100 ADSs (or fraction thereof) cancelled; |
| (iii) | Cash Distribution Fee: by any Holder of ADSs, a fee not in excess of U.S. $5.00 per 100 ADSs (or fraction thereof) held for the distribution of cash dividends or other cash distributions (e.g., upon a sale of rights and other entitlements); |
| (iv) | Stock Distribution /Rights Exercise Fee: by any Holder of ADS(s), a fee not in excess of U.S. $5.00 per 100 ADSs (or fraction thereof) held for the distribution of ADSs pursuant to (a) stock dividends or other free stock distributions, or (b) an exercise of rights to purchase additional ADSs; |
| (v) | Other Distribution Fee: by any Holder of ADS(s), a fee not in excess of U.S. $5.00 per 100 ADSs (or fraction thereof) held for the distribution of securities other than ADSs or rights to purchase additional ADSs (e.g., spin-off shares); |
| (vi) | Depositary Services Fee: by any Holder of ADS(s), a fee not in excess of U.S. $5.00 per 100 ADSs (or fraction thereof) held on the applicable record date(s) established by the Depositary; |
| (vii) | Registration of ADS Transfer Fee: by any Holder of ADS(s) being transferred or by any person to whom ADSs are transferred, a fee not in excess of U.S. $5.00 per 100 ADSs (or fraction thereof) transferred; and |
| (viii) | ADS Conversion Fee: by any Holder of ADS(s) being converted or by any person to whom the converted ADSs are delivered, a fee not in excess of U.S. $5.00 per 100 ADSs (or fraction thereof) converted from one ADS series to another ADS series (e.g., upon conversion of Partial Entitlement ADSs for Full Entitlement ADSs, or upon conversion of Restricted ADSs into freely transferrable ADSs, and vice versa). |
The Company, Holders, Beneficial Owners, persons depositing Shares or withdrawing Deposited Securities in connection with ADS issuances and cancellations, and persons for whom ADSs are issued or cancelled shall be responsible for the following ADS charges under the terms of the Deposit Agreement:
| (a) | taxes (including applicable interest and penalties) and other governmental charges; |
| (b) | such registration fees as may from time to time be in effect for the registration of Shares or other Deposited Securities on the share register and applicable to transfers of Shares or other Deposited Securities to or from the name of the Custodian, the Depositary or any nominees upon the making of deposits and withdrawals, respectively; |
| (c) | such cable, telex and facsimile transmission and delivery expenses as are expressly provided in the Deposit Agreement to be at the expense of the person depositing Shares or withdrawing Deposited Securities or of the Holders and Beneficial Owners of ADSs; |
| (d) | in connection with the conversion of Foreign Currency, the fees, expenses, spreads, taxes and other charges of the Depositary and/or conversion service providers (which may be a division, branch or Affiliate of the Depositary). Such fees, expenses, spreads, taxes and other charges shall be deducted from the Foreign Currency; |
| (e) | any reasonable and customary out-of-pocket expenses incurred in such conversion and/or on behalf of the Holders and Beneficial Owners in complying with currency exchange control or other governmental requirements; and |
| (f) | the fees, charges, costs and expenses incurred by the Depositary, the Custodian, or any nominee in connection with the ADR program. |
All ADS fees and charges may, at any time and from time to time, be changed by agreement between the Depositary and Company but, in the case of ADS fees and charges payable by Holders and Beneficial Owners, only in the manner contemplated by paragraph (23) of this ADR and as contemplated in Section 6.1 of the Deposit Agreement. The Depositary shall provide, without charge, a copy of its latest ADS fee schedule to anyone upon request.
ADS fees and charges payable (i) the issuance of ADSs and (ii) the cancellation of ADSs will be payable by the person for whom the ADSs are so issued by the Depositary (in the case of ADS issuances) and by the person for whom ADSs are being cancelled (in the case of ADS cancellations). In the case of ADSs issued by the Depositary into DTC or presented to the Depositary via DTC, the ADS issuance and cancellation fees and charges will be payable by the DTC Participant(s) receiving the ADSs from the Depositary or the DTC Participant(s) holding the ADSs being cancelled, as the case may be, on behalf of the Beneficial Owner(s) and will be charged by the DTC Participant(s) to the account(s) of the applicable Beneficial Owner(s) in accordance with the procedures and practices of the DTC Participant(s) as in effect at the time. ADS fees and charges in respect of distributions and the ADS service fee are payable by Holders as of the applicable ADS Record Date established by the Depositary. In the case of distributions of cash, the amount of the applicable ADS fees and charges is deducted from the funds being distributed. In the case of (i) distributions other than cash and (ii) the ADS service fee, the applicable Holders as of the ADS Record Date established by the Depositary will be invoiced for the amount of the ADS fees and charges and such ADS fees may be deducted from distributions made to Holders. For ADSs held through DTC, the ADS fees and charges for distributions other than cash and the ADS service fee may be deducted from distributions made through DTC and may be charged to the DTC Participants in accordance with the procedures and practices prescribed by DTC from time to time and the DTC Participants in turn charge the amount of such ADS fees and charges to the Beneficial Owners for whom they hold ADSs. In the case of (i) registration of ADS transfers, the ADS transfer fee will be payable by the ADS Holder whose ADSs are being transferred or by the person to whom the ADSs are transferred, and (ii) conversion of ADSs of one series for ADSs of another series, the ADS conversion fee will be payable by the Holder whose ADSs are converted or by the person to whom the converted ADSs are delivered.
The Depositary may reimburse the Company for certain expenses incurred by the Company in respect of the ADR program established pursuant to the Deposit Agreement, by making available a portion of the ADS fees charged in respect of the ADR program or otherwise, upon such terms and conditions as the Company and the Depositary agree from time to time. The Company shall pay to the Depositary such fees and charges, and reimburse the Depositary for such out-of-pocket expenses, as the Depositary and the Company may agree from time to time. Responsibility for payment of such fees, charges and reimbursements may from time to time be changed by agreement between the Company and the Depositary. Unless otherwise agreed, the Depositary shall present its statement for such fees, charges and reimbursements to the Company once every three months. The charges and expenses of the Custodian are for the sole account of the Depositary.
The obligations of Holders and Beneficial Owners to pay ADS fees and charges shall survive the termination of the Deposit Agreement. As to any Depositary, upon the resignation or removal of such Depositary as described in Section 5.4 of the Deposit Agreement, the right to collect ADS fees and charges shall extend for those ADS fees and charges incurred prior to the effectiveness of such resignation or removal.
(12) Title to ADRs. Subject to the limitations contained in the Deposit Agreement and in this ADR, it is a condition of this ADR, and every successive Holder of this ADR by accepting or holding the same consents and agrees, that title to this ADR (and to each Certificated ADS evidenced hereby) shall be transferable upon the same terms as a certificated security under the laws of the State of New York, provided that, in the case of Certificated ADSs, this ADR has been properly endorsed or is accompanied by proper instruments of transfer. Notwithstanding any notice to the contrary, the Depositary and the Company may deem and treat the Holder of this ADR (that is, the person in whose name this ADR is registered on the books of the Depositary) as the absolute owner thereof for all purposes. Neither the Depositary nor the Company shall have any obligation nor be subject to any liability under the Deposit Agreement or this ADR to any holder of this ADR or any Beneficial Owner unless, in the case of a holder of ADSs, such holder is the Holder of this ADR registered on the books of the Depositary or, in the case of a Beneficial Owner, such Beneficial Owner, or the Beneficial Owner’s representative, is the Holder registered on the books of the Depositary.
(13) Validity of ADR. The Holder(s) of this ADR (and the ADSs represented hereby) shall not be entitled to any benefits under the Deposit Agreement or be valid or enforceable for any purpose against the Depositary or the Company unless this ADR has been (i) dated, (ii) signed by the manual or facsimile signature of a duly-authorized signatory of the Depositary, (iii) countersigned by the manual or facsimile signature of a duly-authorized signatory of the Registrar, and (iv) registered in the books maintained by the Registrar for the registration of issuances and transfers of ADRs. An ADR bearing the facsimile signature of a duly-authorized signatory of the Depositary or the Registrar, who at the time of signature was a duly authorized signatory of the Depositary or the Registrar, as the case may be, shall bind the Depositary, notwithstanding the fact that such signatory has ceased to be so authorized prior to the delivery of such ADR by the Depositary.
(14) Available Information; Reports; Inspection of Transfer Books. The Company is subject to the periodic reporting requirements of the Exchange Act and, accordingly, is required to file or furnish certain reports with the Commission. These reports can be retrieved from the Commission’s website (www.sec.gov) and can be inspected and copied at the public reference facilities maintained by the Commission located (as of the date of the Deposit Agreement) at 100 F Street, N.E., Washington D.C. 20549.
The Depositary shall make available for inspection by Holders at its Principal Office, as promptly as practicable after receipt thereof, any reports and communications, including any proxy soliciting materials, received from the Company which are both (a) received by the Depositary, the Custodian, or the nominee of either of them as the holder of the Deposited Property and (b) made generally available to the holders of such Deposited Property by the Company.
The Registrar shall keep books for the registration of ADSs which at all reasonable times shall be open for inspection by the Company and by the Holders of such ADSs, provided that such inspection shall not be, to the Registrar’s knowledge, for the purpose of communicating with Holders of such ADSs in the interest of a business or object other than the business of the Company or other than a matter related to the Deposit Agreement or the ADSs.
The Registrar may close the transfer books with respect to the ADSs, at any time or from time to time, when deemed necessary or advisable by it in good faith in connection with the performance of its duties hereunder, or at the reasonable written request of the Company subject, in all cases, to paragraph (25) and Section 7.8 of the Deposit Agreement.
Dated:
| CITIBANK, N.A. Transfer Agent and Registrar |
CITIBANK, N.A. as Depositary |
| By: __________________________________ | By: __________________________________ |
| Authorized Signatory | Authorized Signatory |
The address of the Principal Office of the Depositary is 388 Greenwich Street, New York, New York 10013, U.S.A.
[FORM OF REVERSE OF ADR]
SUMMARY OF CERTAIN ADDITIONAL PROVISIONS
OF THE DEPOSIT AGREEMENT
(15) Dividends and Distributions in Cash, Shares, etc. (a) Cash Distributions: Upon the timely receipt by the Depositary of a notice from the Company that it intends to make a distribution of a cash dividend or other cash distribution, the Depositary shall establish an ADS Record Date upon the terms described in Section 4.9 of the Deposit Agreement. Upon receipt of confirmation of receipt of (x) any cash dividend or other cash distribution on any Deposited Securities, or (y) proceeds from the sale of any Deposited Property held in respect of the ADSs under the terms of the Deposit Agreement, the Depositary will (i) if at the time of receipt thereof any amounts are received in a Foreign Currency can, in the judgment of the Depositary (pursuant to Section 4.8 of the Deposit Agreement), be converted on a practical basis into Dollars transferable to the United States, promptly convert or cause to be converted such cash dividend, distribution or proceeds into Dollars (subject to the terms and conditions described in Section 4.8 of the Deposit Agreement), (ii) if applicable and unless previously established, establish the ADS Record Date upon the terms described in Section 4.9 of the Deposit Agreement, and (iii) distribute promptly the amount thus received (net of (a) the applicable fees and charges described in the Fee Schedule attached as Exhibit B to the Deposit Agreement and (b) applicable taxes required to be withheld in connection with the distribution) to the Holders entitled thereto as of the ADS Record Date in proportion to the number of ADSs held as of the ADS Record Date. The Depositary shall distribute only such amount, however, as can be distributed without attributing to any Holder a fraction of one cent, and any balance not so distributed shall be held by the Depositary (without liability for interest thereon) and shall be added to and become part of the next sum received by the Depositary for distribution to Holders of ADSs outstanding at the time of the next distribution. If the Company, the Custodian or the Depositary is required to withhold and does withhold from any cash dividend or other cash distribution in respect of any Deposited Securities, or from any cash proceeds from the sales of Deposited Property, an amount on account of taxes, duties or other governmental charges, the amount distributed to Holders on the ADSs shall be reduced accordingly. Such withheld amounts shall be forwarded by the Company, the Custodian or the Depositary to the relevant governmental authority. Evidence of payment thereof by the Company shall be forwarded by the Company to the Depositary upon request. The Depositary will hold any cash amounts it is unable to distribute in a non-interest bearing account for the benefit of the applicable Holders and Beneficial Owners of ADSs until the distribution can be effected or the funds that the Depositary holds must be escheated as unclaimed property in accordance with the laws of the relevant states of the United States. Notwithstanding anything contained in the Deposit Agreement to the contrary, in the event the Company fails to give the Depositary timely notice of the proposed distribution provided for above, the Depositary agrees to use commercially reasonable efforts to perform the actions contemplated in Section 4.1 of the Deposit Agreement, and the Company, the Holders and the Beneficial Owners acknowledge that the Depositary shall have no liability for the Depositary’s failure to perform the actions contemplated in Section 4.1 of the Deposit Agreement where such notice has not been so timely given, other than its failure to use commercially reasonable efforts, as provided herein.
(b) Share Distributions: Upon the timely receipt by the Depositary of a notice from the Company that it intends to make a distribution that consists of a dividend in, or free distribution of Shares, the Depositary shall establish the ADS Record Date upon the terms described in Section 4.9 of the Deposit Agreement. Upon receipt of confirmation from the Custodian of the receipt of the Shares so distributed by the Company, the Depositary shall either (i) subject to Section 5.9 of the Deposit Agreement, distribute to the Holders as of the ADS Record Date in proportion to the number of ADSs held as of the ADS Record Date, additional ADSs, which represent in the aggregate the number of Shares received as such dividend, or free distribution, subject to the other terms of the Deposit Agreement (including, without limitation, (a) the applicable fees and charges of, and expenses incurred by, the Depositary and (b) applicable taxes required to be withheld), or (ii) if additional ADSs are not so distributed, take all actions necessary so that each ADS issued and outstanding after the ADS Record Date shall, to the extent permissible by law, thenceforth also represent rights and interests in the additional integral number of Shares distributed upon the Deposited Securities represented thereby (net of (a) the applicable fees and charges of, and expenses incurred by, the Depositary, and (b) applicable taxes required to be withheld). In lieu of delivering fractional ADSs, the Depositary shall sell the number of Shares or ADSs, as the case may be, represented by the aggregate of such fractions and distribute the net proceeds upon the terms described in Section 4.1 of the Deposit Agreement.
In the event that the Depositary determines that any distribution in property (including Shares) is subject to any tax or other governmental charges which the Depositary is obligated to withhold, or, if the Company in the fulfillment of its obligations under Section 5.7 of the Deposit Agreement, has furnished an opinion of U.S. counsel determining that Shares must be registered under the Securities Act or other laws in order to be distributed to Holders (and no such registration statement has been declared effective), the Depositary may dispose of all or a portion of such property (including Shares and rights to subscribe therefor) in such amounts and in such manner, including by public or private sale, as the Depositary deems necessary and practicable, and the Depositary shall distribute the net proceeds of any such sale (after deduction of (a) applicable taxes required to be withheld and (b) fees and charges of, and the expenses incurred by, the Depositary) to Holders entitled thereto upon the terms of Section 4.1 of the Deposit Agreement. The Depositary shall hold and/or distribute any unsold balance of such property in accordance with the provisions of the Deposit Agreement. Notwithstanding anything contained in the Deposit Agreement to the contrary, in the event the Company fails to give the Depositary timely notice of the proposed distribution provided for above, the Depositary agrees to use commercially reasonable efforts to perform the actions contemplated in Section 4.2 of the Deposit Agreement, and the Company, the Holders and the Beneficial Owners acknowledge that the Depositary shall have no liability for the Depositary’s failure to perform the actions contemplated in Section 4.2 of the Deposit Agreement where such notice has not been so timely given, other than its failure to use commercially reasonable efforts, as provided herein.
(c) Elective Distributions in Cash or Shares: Upon the timely receipt of a notice indicating that the Company wishes an elective distribution in cash or Shares to be made available to Holders of ADSs upon the terms described in the Deposit Agreement, the Company and the Depositary shall determine in accordance with the Deposit Agreement whether such distribution is lawful and reasonably practicable. The Depositary shall make such elective distribution available to Holders only if (i) the Company shall have timely requested that the elective distribution be made available to Holders, (ii) the Depositary shall have determined that such distribution is reasonably practicable and (iii) the Depositary shall have received satisfactory documentation within the terms of Section 5.7 of the Deposit Agreement. If the above conditions are satisfied, the Depositary shall, subject to the terms and conditions of the Deposit Agreement, establish the ADS Record Date according to paragraph (16) and establish procedures to enable the Holder hereof to elect to receive the proposed distribution in cash or in additional ADSs. If a Holder elects to receive the distribution in cash, the distribution shall be made as in the case of a distribution in cash. If the Holder hereof elects to receive the distribution in additional ADSs, the distribution shall be made as in the case of a distribution in Shares upon the terms described in the Deposit Agreement. If such elective distribution is not reasonably practicable or if the Depositary did not receive satisfactory documentation set forth in the Deposit Agreement, the Depositary shall establish an ADS Record Date upon the terms of Section 4.9 of the Deposit Agreement and, to the extent permitted by law, distribute to Holders, on the basis of the same determination as is made in England and Wales in respect of the Shares for which no election is made, either (x) cash or (y) additional ADSs representing such additional Shares, in each case, upon the terms described in the Deposit Agreement. Nothing herein or in the Deposit Agreement shall obligate the Depositary to make available to the Holder hereof a method to receive the elective distribution in Shares (rather than ADSs). There can be no assurance that the Holder hereof will be given the opportunity to receive elective distributions on the same terms and conditions as the holders of Shares. Notwithstanding anything contained in the Deposit Agreement to the contrary, in the event the Company fails to give the Depositary timely notice of the proposed distribution provided for above, the Depositary agrees to use commercially reasonable efforts to perform the actions contemplated in Section 4.3 of the Deposit Agreement, and the Company, the Holders and the Beneficial Owners acknowledge that the Depositary shall have no liability for the Depositary’s failure to perform the actions contemplated in Section 4.3 of the Deposit Agreement where such notice has not been so timely given, other than its failure to use commercially reasonable efforts, as provided herein.
(d) Distribution of Rights to Purchase Additional ADSs: Upon the timely receipt by the Depositary of a notice indicating that the Company wishes rights to subscribe for additional Shares to be made available to Holders of ADSs, the Depositary upon consultation with the Company, shall determine, whether it is lawful and reasonably practicable to make such rights available to the Holders. The Depositary shall make such rights available to any Holders only if (i) the Company shall have timely requested that such rights be made available to Holders, (ii) the Depositary shall have received satisfactory documentation within the terms of Section 5.7 of the Deposit Agreement, and (iii) the Depositary shall have determined that such distribution of rights is reasonably practicable. If such conditions are not satisfied or if the Company requests that the rights not be made available to Holders of ADSs, the Depositary shall sell the rights as described below. In the event all conditions set forth above are satisfied, the Depositary shall establish the ADS Record Date (upon the terms described in Section 4.9 of the Deposit Agreement) and establish procedures to (x) distribute rights to purchase additional ADSs (by means of warrants or otherwise), (y) enable the Holders to exercise such rights (upon payment of the subscription price and of the applicable (a) fees and charges of, and expenses incurred by, the Depositary and (b) taxes), and (z) deliver ADSs upon the valid exercise of such rights. Nothing herein or in the Deposit Agreement shall obligate the Depositary to make available to the Holders a method to exercise rights to subscribe for Shares (rather than ADSs). If (i) the Company does not timely request the Depositary to make the rights available to Holders or requests that the rights not be made available to Holders, (ii) the Depositary fails to receive satisfactory documentation within the terms of Section 5.7 of the Deposit Agreement or determines it is not reasonably practicable to make the rights available to Holders, or (iii) any rights made available are not exercised and appear to be about to lapse, the Depositary shall determine whether it is lawful and reasonably practicable to sell such rights, in a riskless principal capacity, at such place and upon such terms (including public and private sale) as it may deem practicable. The Depositary shall, upon such sale, convert and distribute proceeds of such sale (net of applicable (a) fees and charges of, and expenses incurred by, the Depositary and (b) taxes) upon the terms hereof and of Section 4.1 of the Deposit Agreement. If the Depositary is unable to make any rights available to Holders upon the terms described in Section 4.4(a) of the Deposit Agreement or to arrange for the sale of the rights upon the terms described in Section 4.4(b) of the Deposit Agreement, the Depositary shall allow such rights to lapse. The Depositary shall not be liable for (i) any failure to accurately determine whether it may be lawful or practicable to make such rights available to Holders in general or any Holders in particular, (ii) any foreign exchange exposure or loss incurred in connection with such sale or exercise, or (iii) the content of any materials forwarded to the Holders on behalf of the Company in connection with the rights distribution.
Notwithstanding anything herein or in the Deposit Agreement to the contrary, if registration (under the Securities Act or any other applicable law) of the rights or the securities to which any rights relate may be required in order for the Company to offer such rights or such securities to Holders and to sell the securities represented by such rights, the Depositary will not distribute such rights to the Holders (i) unless and until a registration statement under the Securities Act (or other applicable law) covering such offering is in effect or (ii) unless the Company furnishes the Depositary opinion(s) of counsel for the Company in the United States and counsel to the Company in any other applicable country in which rights would be distributed, in each case satisfactory to the Depositary, to the effect that the offering and sale of such securities to Holders and Beneficial Owners are exempt from, or do not require registration under, the provisions of the Securities Act or any other applicable laws. In the event that the Company, the Depositary or the Custodian shall be required to withhold and does withhold from any distribution of Deposited Property (including rights) an amount on account of taxes or other governmental charges, the amount distributed to the Holders of ADSs shall be reduced accordingly. In the event that the Depositary determines that any distribution of Deposited Property (including Shares and rights to subscribe therefor) is subject to any tax or other governmental charges which the Depositary is obligated to withhold, the Depositary may dispose of all or a portion of such Deposited Property (including Shares and rights to subscribe therefor) in such amounts and in such manner, including by public or private sale, as the Depositary deems necessary and practicable to pay any such taxes or charges.
There can be no assurance that Holders generally, or any Holder in particular, will be given the opportunity to receive or exercise rights on the same terms and conditions as the holders of Shares or be able to exercise such rights. Nothing herein or in the Deposit Agreement shall obligate the Company to file any registration statement in respect of any rights or Shares or other securities to be acquired upon the exercise of such rights.
(e) Distributions other than Cash, Shares or Rights to Purchase Shares: Upon receipt of a notice indicating that the Company wishes property other than cash, Shares or rights to purchase additional Shares to be made to Holders of ADSs, the Depositary shall determine whether such distribution to Holders is lawful and reasonably practicable. The Depositary shall not make such distribution unless (i) the Company shall have requested the Depositary to make such distribution to Holders, (ii) the Depositary shall have received the documentation contemplated in the Deposit Agreement, and (iii) the Depositary shall have determined that such distribution is reasonably practicable. Upon satisfaction of such conditions, the Depositary shall distribute the property so received to the Holders of record, as of the ADS Record Date, in proportion to the number of ADSs held by them respectively and in such manner as the Depositary may deem practicable for accomplishing such distribution (i) upon receipt of payment or net of the applicable fees and charges of, and expenses incurred by, the Depositary, and (ii) net of any applicable taxes required to be withheld. The Depositary may dispose of all or a portion of the property so distributed and deposited, in such amounts and in such manner (including public or private sale) as the Depositary may deem practicable or necessary to satisfy any taxes (including applicable interest and penalties) or other governmental charges applicable to the distribution.
If the conditions above are not satisfied, the Depositary shall sell or cause such property to be sold in a public or private sale, at such place or places and upon such terms as it may deem practicable and shall (i) cause the proceeds of such sale, if any, to be converted into Dollars and (ii) distribute the proceeds of such conversion received by the Depositary (net of applicable (a) fees and charges of, and expenses incurred by, the Depositary and (b) taxes) to the Holders as of the ADS Record Date upon the terms hereof and of the Deposit Agreement. If the Depositary is unable to sell such property, the Depositary may dispose of such property for the account of the Holders in any way it deems reasonably practicable under the circumstances.
Neither the Depositary nor the Company shall be responsible for (i) any failure to determine whether it is lawful or practicable to make the property described in Section 4.5 of the Deposit Agreement available to Holders in general or any Holders in particular, nor (ii) any loss incurred in connection with the sale or disposal of such property.
(16) Redemption. Upon timely receipt of notice from the Company that it intends to exercise its right of redemption in respect of any of the Deposited Securities, and satisfactory documentation, and only if after consultation between the Depositary and the Company, the Depositary has determined that such proposed redemption is practicable, the Depositary shall (to the extent practicable) provide to each Holder a notice setting forth the Company’s intention to exercise the redemption rights and any other particulars set forth in the Company’s notice to the Depositary. The Depositary shall instruct the Custodian to present to the Company the Deposited Securities in respect of which redemption rights are being exercised against payment of the applicable redemption price. Upon receipt of confirmation from the Custodian that the redemption has taken place and that funds representing the redemption price have been received, the Depositary shall convert, transfer, and distribute the proceeds (net of applicable (a) fees and charges of, and the expenses incurred by, the Depositary, and (b) taxes), retire ADSs and cancel ADRs, if applicable, upon delivery of such ADSs by Holders thereof and the terms set forth in Sections 4.1 and 6.2 of the Deposit Agreement. If less than all outstanding Deposited Securities are redeemed, the ADSs to be retired will be selected by lot or on a pro rata basis, as may be determined by the Depositary after consultation with the Company. The redemption price per ADS shall be the dollar equivalent of the per share amount received by the Depositary (adjusted to reflect the ADS(s)-to-Share(s) ratio) upon the redemption of the Deposited Securities represented by ADSs (subject to the terms of Section 4.8 of the Deposit Agreement and the applicable fees and charges of, and expenses incurred by, the Depositary, and taxes) multiplied by the number of Deposited Securities represented by each ADS redeemed. Notwithstanding anything contained in the Deposit Agreement to the contrary, in the event the Company fails to give the Depositary timely notice of the proposed redemption provided for above, the Depositary agrees to use commercially reasonable efforts to perform the actions contemplated in Section 4.7 of the Deposit Agreement, and the Company, the Holders and the Beneficial Owners acknowledge that the Depositary shall have no liability for the Depositary’s failure to perform the actions contemplated in Section 4.7 of the Deposit Agreement where such notice has not been so timely given, other than its failure to use commercially reasonable efforts, as provided herein.
(17) Fixing of ADS Record Date. Whenever the Depositary shall receive notice of the fixing of a record date by the Company for the determination of holders of Deposited Securities entitled to receive any distribution (whether in cash, Shares, rights or other distribution), or whenever for any reason the Depositary causes a change in the number of Shares that are represented by each ADS, or whenever the Depositary shall receive notice of any meeting of, or solicitation of consents or proxies of, holders of Shares or other Deposited Securities, or whenever the Depositary shall find it necessary or convenient in connection with the giving of any notice, solicitation of any consent or any other matter, the Depositary shall fix the record date (the “ADS Record Date”) for the determination of the Holders of ADS(s) who shall be entitled to receive such distribution, to give instructions for the exercise of voting rights at any such meeting, to give or withhold such consent, to receive such notice or solicitation or to otherwise take action, or to exercise the rights of Holders with respect to such changed number of Shares represented by each ADS. Subject to applicable law, the terms and conditions of this ADR and Sections 4.1 through 4.8 of the Deposit Agreement, only the Holders of ADSs at the close of business in New York on such ADS Record Date shall be entitled to receive such distribution, to give such voting instructions, to receive such notice or solicitation, or otherwise take action.
(18) Voting of Deposited Securities. As soon as practicable after receipt of notice of any meeting at which the holders of Deposited Securities are entitled to vote, or of solicitation of consents or proxies from holders of Deposited Securities, the Depositary shall fix the ADS Record Date in respect of such meeting or solicitation of consent or proxy in accordance with Section 4.9 of the Deposit Agreement. The Depositary shall, if requested by the Company in writing in a timely manner (the Depositary having no obligation to take any further action if the request shall not have been received by the Depositary at least thirty (30) days prior to the date of such vote or meeting), at the Company’s expense and provided no U.S. legal prohibitions exist, distribute to Holders as of the ADS Record Date: (a) such notice of meeting or solicitation of consent or proxy, (b) a statement that the Holders at the close of business on the ADS Record Date will be entitled, subject to any applicable law, the provisions of the Deposit Agreement, the Articles of Association of the Company and the provisions of or governing the Deposited Securities (which provisions, if any, shall be summarized in pertinent part by the Company), to instruct the Depositary as to the exercise of the voting rights, if any, pertaining to the Deposited Securities represented by such Holder’s ADSs, and (c) a brief statement as to the manner in which such voting instructions may be given to the Depositary or in which voting instructions may be deemed to have been given in accordance with this Section 4.10 of the Deposit Agreement.
Notwithstanding anything contained in the Deposit Agreement or any ADR, with the Company’s prior written consent, the Depositary may, to the extent not prohibited by law or regulations, or by the requirements of any stock exchange on which the ADSs may be listed, in lieu of distribution of the materials provided to the Depositary in connection with any meeting of, or solicitation of consents or proxies from, holders of Deposited Securities, distribute to the Holders a notice that provides Holders with, or otherwise publicizes to Holders, instructions on how to retrieve such materials or receive such materials upon request (e.g., by reference to a website containing the materials for retrieval or a contact for requesting copies of the materials).
The Depositary has been advised by the Company that under the Articles of Association of the Company as in effect on the date of the Deposit Agreement, voting at any meeting of shareholders of the Company is by poll.
Voting instructions may be given only in respect of a number of ADSs representing an integral number of Deposited Securities. Upon the timely receipt from a Holder of ADSs as of the ADS Record Date of voting instructions in the manner specified by the Depositary, the Depositary shall endeavor, insofar as practicable and permitted under any applicable law, the provisions of the Deposit Agreement, the Articles of Association of the Company and the provisions of the Deposited Securities, to vote, or cause the Custodian to vote, the Deposited Securities (in person or by proxy) represented by such Holder’s ADSs in accordance with the voting instructions received from the Holder of the ADSs. If the Depositary does not receive voting instructions from a Holder as of the ADS Record Date on or before the date established by the Depositary for such purpose, such Holder shall be deemed, and the Depositary shall deem such Holder, to have instructed the Depositary to give a discretionary proxy to a person designated by the Company to vote the Deposited Securities; provided, however, that no such discretionary proxy shall be given by the Depositary with respect to any matter to be voted upon as to which the Company informs the Depositary that (i) the Company does not wish such proxy to be given, (ii) substantial opposition exists, or (iii) the rights of holders of Deposited Securities may be adversely affected.
Deposited Securities represented by ADSs for which no timely voting instructions are received by the Depositary from the Holder shall not be voted (except as otherwise contemplated herein). Neither the Depositary nor the Custodian shall under any circumstances exercise any discretion as to voting and neither the Depositary nor the Custodian shall vote, attempt to exercise the right to vote, or in any way make use of, for purposes of establishing a quorum or otherwise, the Deposited Securities represented by ADSs, except pursuant to and in accordance with the voting instructions timely received from Holders or as otherwise contemplated in the Deposit Agreement or herein. If the Depositary timely receives voting instructions from a Holder which fail to specify the manner in which the Depositary is to vote the Deposited Securities represented by such Holder’s ADSs, the Depositary will deem such Holder (unless otherwise specified in the notice distributed to Holders) to have instructed the Depositary to vote in favor of the items set forth in such voting instructions.
Notwithstanding anything else contained herein, the Depositary shall, if so requested in writing by the Company, represent all Deposited Securities (whether or not voting instructions have been received in respect of such Deposited Securities from Holders as of the ADS Record Date) for the sole purpose of establishing quorum at a meeting of shareholders.
Notwithstanding anything else contained in the Deposit Agreement or this ADR, the Depositary shall not have any obligation to take any action with respect to any meeting, or solicitation of consents or proxies, of holders of Deposited Securities if the taking of such action would violate the laws of the United States or England & Wales. The Company agrees to take any and all actions reasonably necessary and as permitted by the laws of England and Wales to enable Holders and Beneficial Owners to exercise the voting rights accruing to the Deposited Securities and to deliver to the Depositary an opinion of U.S. counsel addressing any actions requested to be taken if so requested by the Depositary. There can be no assurance that Holders generally or any Holder in particular will receive the notice described above with sufficient time to enable the Holder to return voting instructions to the Depositary in a timely manner.
(19) Changes Affecting Deposited Securities. Upon any change in nominal value, sub-division, cancellation, consolidation or any other reclassification of Deposited Securities, or upon any recapitalization, reorganization, merger, scheme of arrangement, consolidation or sale of assets affecting the Company or to which it is a party, any property which shall be received by the Depositary or the Custodian in exchange for, or in conversion of, or replacement of, or otherwise in respect of, such Deposited Securities shall, to the extent permitted by law, be treated as new Deposited Property under the Deposit Agreement, and this ADR shall, subject to the provisions of the Deposit Agreement, this ADR evidencing such ADSs and applicable law, represent the right to receive such additional or replacement Deposited Property. In giving effect to such change, sub-division, cancellation, consolidation or other reclassification of Deposited Securities, recapitalization, reorganization, merger, scheme of arrangement, consolidation or sale of assets, the Depositary may, with the Company’s approval, and shall, if the Company shall so request, subject to the terms of the Deposit Agreement (including, without limitation, (a) the applicable fees and charges of, and expenses incurred by, the Depositary, and (b) applicable taxes) and receipt of an opinion of counsel to the Company satisfactory to the Depositary that such actions are not in violation of any applicable laws or regulations, (i) issue and deliver additional ADSs as in the case of a stock dividend on the Shares, (ii) amend the Deposit Agreement and the applicable ADRs, (iii) amend the applicable Registration Statement(s) on Form F-6 as filed with the Commission in respect of the ADSs, (iv) call for the surrender of outstanding ADRs to be exchanged for new ADRs, and (v) take such other actions as are appropriate to reflect the transaction with respect to the ADSs. Notwithstanding the foregoing, in the event that any Deposited Property so received may not be lawfully distributed to some or all Holders, the Depositary may, with the Company’s approval, and shall, if the Company requests, subject to receipt of an opinion of Company’s counsel satisfactory to the Depositary that such action is not in violation of any applicable laws or regulations, sell such Deposited Property at public or private sale, at such place or places and upon such terms as it may deem proper and may allocate the net proceeds of such sales (net of (a) fees and charges of, and expenses incurred by, the Depositary and (b) taxes) for the account of the Holders otherwise entitled to such Deposited Property upon an averaged or other practicable basis without regard to any distinctions among such Holders and distribute the net proceeds so allocated to the extent practicable as in the case of a distribution received in cash pursuant to Section 4.1 of the Deposit Agreement. The Depositary shall not be responsible for (i) any failure to determine that it may be lawful or practicable to make such Deposited Property available to Holders in general or to any Holder in particular, (ii) any foreign exchange exposure or loss incurred in connection with such sale, or (iii) any liability to the purchaser of such Deposited Property.
(20) Exoneration. Notwithstanding anything contained in the Deposit Agreement or any ADR, neither the Depositary nor the Company shall be obligated to do or perform any act which is inconsistent with the provisions of the Deposit Agreement or incur any liability (to the extent not limited by paragraph (25) hereof) (i) if the Depositary, the Custodian, the Company or their respective agents shall be prevented or forbidden from, or delayed in, doing or performing any act or thing required or contemplated by the terms of the Deposit Agreement and this ADR, by reason of any provision of any present or future law or regulation of the United States, England and Wales, or any other country, or of any other governmental authority or regulatory authority or stock exchange, or on account of potential criminal or civil penalties or restraint, or by reason of any provision, present or future, of the Articles of Association of the Company or any provision of or governing any Deposited Securities, or by reason of any act of God or war or other circumstances beyond its control (including, without limitation, nationalization, expropriation, currency restrictions, work stoppage, strikes, civil unrest, acts of terrorism, revolutions, rebellions, explosions and computer failure), (ii) by reason of any exercise of, or failure to exercise, any discretion provided for in the Deposit Agreement or in the Articles of Association of the Company or provisions of or governing Deposited Securities, (iii) for any action or inaction in reliance upon the advice of or information from legal counsel, accountants, any person presenting Shares for deposit, any Holder, any Beneficial Owner or authorized representative thereof, or any other person believed by it in good faith to be competent to give such advice or information, (iv) for the inability by a Holder or Beneficial Owner to benefit from any distribution, offering, right or other benefit which is made available to holders of Deposited Securities but is not, under the terms of the Deposit Agreement, made available to Holders of ADSs, (v) for any action or inaction of any clearing or settlement system (any participant thereof) for the Deposited Property or the ADSs, or (vi) for any consequential or punitive damages (including lost profits) for any breach of the terms of the Deposit Agreement. The Depositary, its controlling persons, its agents, any Custodian and the Company, its controlling persons and its agents may rely and shall be protected in acting upon any written notice, request or other document believed by it to be genuine and to have been signed or presented by the proper party or parties.
(21) Standard of Care. The Company and the Depositary assume no obligation and shall not be subject to any liability under the Deposit Agreement or this ADR to any Holder(s) or Beneficial Owner(s), except that the Company and the Depositary agree to perform their respective obligations specifically set forth in the Deposit Agreement or this ADR without negligence or bad faith. Without limitation of the foregoing, neither the Depositary, nor the Company, nor any of their respective controlling persons, or agents, shall be under any obligation to appear in, prosecute or defend any action, suit or other proceeding in respect of any Deposited Property or in respect of the ADSs, which in its opinion may involve it in expense or liability, unless indemnity satisfactory to it against all expense (including fees and disbursements of counsel) and liability be furnished as often as may be required (and no Custodian shall be under any obligation whatsoever with respect to such proceedings, the responsibility of the Custodian being solely to the Depositary).
The Depositary and its agents shall not be liable for any failure to carry out any instructions to vote any of the Deposited Securities, or for the manner in which any vote is cast or the effect of any vote, provided that any such action or omission is in good faith and without negligence and in accordance with the terms of the Deposit Agreement. The Depositary shall not incur any liability for any failure to accurately determine that any distribution or action may be lawful or reasonably practicable, for the content of any information submitted to it by the Company for distribution to the Holders or for any inaccuracy of any translation thereof, for any investment risk associated with acquiring an interest in the Deposited Property, for the validity or worth of the Deposited Property or for any tax consequences that may result from the ownership of ADSs, Shares or other Deposited Property, for the credit-worthiness of any third party, for allowing any rights to lapse upon the terms of the Deposit Agreement, for the failure or timeliness of any notice from the Company, or for any action of or failure to act by, or any information provided or not provided by, DTC or any DTC Participant.
The Depositary shall not be liable for any acts or omissions made by a successor depositary in connection with any matter arising wholly prior to the appointment of the Depositary or after the removal or resignation of the Depositary, provided that in connection with the issue out of which such potential liability arises, the Depositary performed its obligations without negligence or bad faith while it acted as Depositary for the Company.
(22) Resignation and Removal of the Depositary; Appointment of Successor Depositary. The Depositary may at any time resign as Depositary under the Deposit Agreement by written notice of resignation delivered to the Company, such resignation to be effective on the earlier of (i) the 90th day after delivery thereof to the Company (whereupon the Depositary shall be entitled to take the actions contemplated in Section 6.2 of the Deposit Agreement), or (ii) the appointment by the Company of a successor depositary and its acceptance of such appointment as provided in the Deposit Agreement. The Depositary may at any time be removed by the Company by written notice of such removal, which removal shall be effective on the later of (i) the 90th day after delivery thereof to the Depositary (whereupon the Depositary shall be entitled to take the actions contemplated in Section 6.2 of the Deposit Agreement), or (ii) upon the appointment by the Company of a successor depositary and its acceptance of such appointment as provided in the Deposit Agreement. In case at any time the Depositary acting hereunder shall resign or be removed, the Company shall use its best efforts to appoint a successor depositary, which shall be a bank or trust company having an office in the Borough of Manhattan, the City of New York. Every successor depositary shall be required by the Company to execute and deliver to its predecessor and to the Company an instrument in writing accepting its appointment hereunder, and thereupon such successor depositary, without any further act or deed (except as required by applicable law), shall become fully vested with all the rights, powers, duties and obligations of its predecessor (other than as contemplated in Sections 5.8 and 5.9 of the Deposit Agreement). The predecessor depositary, upon payment of all sums due it and on the written request of the Company shall (i) execute and deliver an instrument transferring to such successor all rights and powers of such predecessor hereunder (other than as contemplated in Sections 5.8 and 5.9 of the Deposit Agreement), (ii) duly assign, transfer and deliver all of the Depositary’s right, title and interest to the Deposited Property to such successor, and (iii) deliver to such successor a list of the Holders of all outstanding ADSs and such other information relating to ADSs and Holders thereof as the successor may reasonably request. Any such successor depositary shall promptly provide notice of its appointment to such Holders. Any entity into or with which the Depositary may be merged or consolidated shall be the successor of the Depositary without the execution or filing of any document or any further act.
(23) Amendment/Supplement. Subject to the terms and conditions of this paragraph 23, and Section 6.1 of the Deposit Agreement and applicable law, this ADR and any provisions of the Deposit Agreement may at any time and from time to time be amended or supplemented by written agreement between the Company and the Depositary in any respect which they may deem necessary or desirable without the prior written consent of the Holders or Beneficial Owners. Any amendment or supplement which shall impose or increase any fees or charges (other than charges in connection with foreign exchange control regulations, and taxes and other governmental charges, delivery and other such expenses), or which shall otherwise materially prejudice any substantial existing right of Holders or Beneficial Owners, shall not, however, become effective as to outstanding ADSs until the expiration of thirty (30) days after notice of such amendment or supplement shall have been given to the Holders of outstanding ADSs. Notice of any amendment to the Deposit Agreement or any ADR shall not need to describe in detail the specific amendments effectuated thereby, and failure to describe the specific amendments in any such notice shall not render such notice invalid, provided, however, that, in each such case, the notice given to the Holders identifies a means for Holders and Beneficial Owners to retrieve or receive the text of such amendment (e.g., upon retrieval from the Commission’s, the Depositary’s or the Company’s website or upon request from the Depositary). The parties hereto agree that any amendments or supplements which (i) are reasonably necessary (as agreed by the Company and the Depositary) in order for (a) the ADSs to be registered on Form F-6 under the Securities Act or (b) the ADSs to be settled solely in electronic book-entry form and (ii) do not in either such case impose or increase any fees or charges to be borne by Holders, shall be deemed not to materially prejudice any substantial existing rights of Holders or Beneficial Owners. Every Holder and Beneficial Owner at the time any amendment or supplement so becomes effective shall be deemed, by continuing to hold such ADSs, to consent and agree to such amendment or supplement and to be bound by the Deposit Agreement and this ADR, if applicable, as amended or supplemented thereby. In no event shall any amendment or supplement impair the right of the Holder to surrender such ADS and receive therefor the Deposited Securities represented thereby, except in order to comply with mandatory provisions of applicable law. Notwithstanding the foregoing, if any governmental body should adopt new laws, rules or regulations which would require an amendment of, or supplement to, the Deposit Agreement to ensure compliance therewith, the Company and the Depositary may amend or supplement the Deposit Agreement and this ADR at any time in accordance with such changed laws, rules or regulations. Such amendment or supplement to the Deposit Agreement and this ADR in such circumstances may become effective before a notice of such amendment or supplement is given to Holders or within any other period of time as required for compliance with such laws, rules or regulations.
(24) Termination. The Depositary shall, at any time at the written direction of the Company, terminate the Deposit Agreement by distributing notice of such termination to the Holders of all ADSs then outstanding at least thirty (30) days prior to the date fixed in such notice for such termination. If (i) ninety (90) days shall have expired after the Depositary shall have delivered to the Company a written notice of its election to resign, or (ii) ninety (90) days shall have expired after the Company shall have delivered to the Depositary a written notice of the removal of the Depositary, and, in either case, a successor depositary shall not have been appointed and accepted its appointment as provided in Section 5.4 of the Deposit Agreement, the Depositary may terminate the Deposit Agreement by distributing notice of such termination to the Holders of all ADSs then outstanding at least thirty (30) days prior to the date fixed in such notice for such termination. The date so fixed for termination of the Deposit Agreement in any termination notice so distributed by the Depositary to the Holders of ADSs is referred to as the “Termination Date”. Until the Termination Date, the Depositary shall continue to perform all of its obligations under the Deposit Agreement, and the Holders and Beneficial Owners will be entitled to all of their rights under the Deposit Agreement. If any ADSs shall remain outstanding after the Termination Date, the Registrar and the Depositary shall not, after the Termination Date, have any obligation to perform any further acts under the Deposit Agreement, except that the Depositary shall, subject, in each case, to the terms and conditions of the Deposit Agreement, continue to (i) collect dividends and other distributions pertaining to Deposited Securities, (ii) sell Deposited Property received in respect of Deposited Securities, (iii) deliver Deposited Securities, together with any dividends or other distributions received with respect thereto and the net proceeds of the sale of any other Deposited Property, in exchange for ADSs surrendered to the Depositary (after deducting, or charging, as the case may be, in each case, the fees and charges of, and expenses incurred by, the Depositary, and all applicable taxes or governmental charges for the account of the Holders and Beneficial Owners, in each case upon the terms set forth in Section 5.9 of the Deposit Agreement), and (iv) take such actions as may be required under applicable law in connection with its role as Depositary under the Deposit Agreement. At any time after the Termination Date, the Depositary may sell the Deposited Property then held under the Deposit Agreement and shall after such sale hold un-invested the net proceeds of such sale, together with any other cash then held by it under the Deposit Agreement, in an un-segregated account and without liability for interest, for the pro rata benefit of the Holders whose ADSs have not theretofore been surrendered. After making such sale, the Depositary shall be discharged from all obligations under the Deposit Agreement except (i) to account for such net proceeds and other cash (after deducting, or charging, as the case may be, in each case, the fees and charges of, and expenses incurred by, the Depositary, and all applicable taxes or governmental charges for the account of the Holders and Beneficial Owners, in each case upon the terms set forth in Section 5.9 of the Deposit Agreement), and (ii) as may be required at law in connection with the termination of the Deposit Agreement. After the Termination Date, the Company shall be discharged from all obligations under the Deposit Agreement, except for its obligations to the Depositary under Sections 5.8, 5.9 and 7.6 of the Deposit Agreement. The obligations under the terms of the Deposit Agreement of Holders and Beneficial Owners of ADSs outstanding as of the Termination Date shall survive the Termination Date and shall be discharged only when the applicable ADSs are presented by their Holders to the Depositary for cancellation under the terms of the Deposit Agreement (except as specifically provided in the Deposit Agreement).
Notwithstanding anything contained in the Deposit Agreement or this ADR, in connection with the termination of the Deposit Agreement, the Depositary may, independently and without the need for any action by the Company, make available to Holders of ADSs a means to withdraw the Deposited Securities represented by their ADSs and to direct the deposit of such Deposited Securities into an unsponsored American depositary shares program established by the Depositary, upon such terms and conditions as the Depositary may deem reasonably appropriate, subject however, in each case, to satisfaction of the applicable registration requirements by the unsponsored American depositary shares program under the Securities Act, and to receipt by the Depositary of payment of the applicable fees and charges of, and reimbursement of the applicable expenses incurred by, the Depositary.
(25) Compliance with, and No Disclaimer under, U.S. Securities Laws. (a) Notwithstanding any provisions in this ADR or the Deposit Agreement to the contrary, the withdrawal or delivery of Deposited Securities will not be suspended by the Company or the Depositary except as would be permitted by Instruction I.A.(1) of the General Instructions to the Form F-6 Registration Statement, as amended from time to time, under the Securities Act.
(b) Each of the parties to the Deposit Agreement (including, without limitation, each Holder and Beneficial Owner) acknowledges and agrees that no provision of the Deposit Agreement or any ADR shall, or shall be deemed to, disclaim any liability under the Securities Act or the Exchange Act, in each case to the extent established under applicable U.S. laws.
(26) No Third Party Beneficiaries / Acknowledgements. The Deposit Agreement is for the exclusive benefit of the parties hereto (and their successors) and shall not be deemed to give any legal or equitable right, remedy or claim whatsoever to any other person, except to the extent specifically set forth in the Deposit Agreement. Nothing in the Deposit Agreement shall be deemed to give rise to a partnership or joint venture among the parties nor establish a fiduciary or similar relationship among the parties. The parties hereto acknowledge and agree that (i) Citibank and its Affiliates may at any time have multiple banking relationships with the Company, the Holders, the Beneficial Owners, and their respective Affiliates, (ii) Citibank and its Affiliates may own and deal in any class of securities of the Company and its Affiliates and in ADSs, and may be engaged at any time in transactions in which parties adverse to the Company, the Holders, the Beneficial Owners or their respective Affiliates may have interests, (iii) the Depositary and its Affiliates may from time to time have in their possession non-public information about the Company, the Holders, the Beneficial Owners, and their respective Affiliates, (iv) nothing contained in the Deposit Agreement shall (a) preclude Citibank or any of its Affiliates from engaging in such transactions or establishing or maintaining such relationships, or (b) obligate Citibank or any of its Affiliates to disclose such information, transactions or relationships, or to account for any profit made or payment received in such transactions or relationships, (v) the Depositary shall not be deemed to have knowledge of any information any other division of Citibank or any of its Affiliates may have about the Company, the Holders, the Beneficial Owners, or any of their respective Affiliates, and (vi) the Company, the Depositary, the Custodian and their respective agents and controlling persons may be subject to the laws and regulations of jurisdictions other than the United States and England and Wales, and the authority of courts and regulatory authorities of such other jurisdictions, and, consequently, the requirements and the limitations of such other laws and regulations, and the decisions and orders of such other courts and regulatory authorities, may affect the rights and obligations of the parties to the Deposit Agreement.
(27) Governing Law / Waiver of Jury Trial. The Deposit Agreement, the ADRs, and the ADSs shall be interpreted in accordance with, and all rights hereunder and thereunder and provisions hereof and thereof shall be governed by, the laws of the State of New York applicable to contracts made and to be wholly performed in that State. Notwithstanding anything contained in the Deposit Agreement to the contrary, any ADR or any present or future provisions of the laws of the State of New York, the rights of holders of Shares and of any other Deposited Securities and the obligations and duties of the Company in respect of the holders of Shares and other Deposited Securities, as such, shall be governed by the laws of England and Wales (or, if applicable, such other laws as may govern the Deposited Securities).
EACH OF THE PARTIES TO THE DEPOSIT AGREEMENT (INCLUDING, WITHOUT LIMITATION, EACH HOLDER AND BENEFICIAL OWNER) IRREVOCABLY WAIVES, TO THE FULLEST EXTENT PERMITTED BY APPLICABLE LAW, ANY AND ALL RIGHT TO TRIAL BY JURY IN ANY LEGAL PROCEEDING AGAINST THE COMPANY AND/OR THE DEPOSITARY ARISING OUT OF, OR RELATING TO, THE DEPOSIT AGREEMENT, ANY ADR AND ANY TRANSACTIONS CONTEMPLATED THEREIN (WHETHER BASED ON CONTRACT, TORT, COMMON LAW OR OTHERWISE).
<
(ASSIGNMENT AND TRANSFER SIGNATURE LINES)
FOR VALUE RECEIVED, the undersigned Holder hereby sell(s), assign(s) and transfer(s) unto ______________________________ whose taxpayer identification number is _______________________ and whose address including postal zip code is ________________, the within ADR and all rights thereunder, hereby irrevocably constituting and appointing ________________________ attorney-in-fact to transfer said ADR on the books of the Depositary with full power of substitution in the premises.
| Dated: | Name: ________________________________ |
| By: | |
| Title: | |
| NOTICE: The signature of the Holder to this assignment must correspond with the name as written upon the face of the within instrument in every particular, without alteration or enlargement or any change whatsoever. | |
| If the endorsement be executed by an attorney, executor, administrator, trustee or guardian, the person executing the endorsement must give his/her full title in such capacity and proper evidence of authority to act in such capacity, if not on file with the Depositary, must be forwarded with this ADR. | |
| __________________________ | |
| SIGNATURE GUARANTEED | |
| All endorsements or assignments of ADRs must be guaranteed by a member of a Medallion Signature Program approved by the Securities Transfer Association, Inc. |
Legends
[The ADRs issued in respect of Partial Entitlement American Depositary Shares shall bear the following legend on the face of the ADR: “This ADR evidences ADSs representing 'partial entitlement' Shares of the Company and as such do not entitle the holders thereof to the same per-share entitlement as other Shares (which are 'full entitlement' Shares) issued and outstanding at such time. The ADSs represented by this ADR shall entitle holders to distributions and entitlements identical to other ADSs when the Shares represented by such ADSs become 'full entitlement' Shares.”]
EXHIBIT B
FEE SCHEDULE
ADS FEES AND RELATED CHARGES
All capitalized terms used but not otherwise defined herein shall have the meaning given to such terms in the Deposit Agreement. Except as otherwise specified herein, any reference to ADSs herein includes Partial Entitlement ADSs, Full Entitlement ADSs, Certificated ADSs, Uncertificated ADSs, and Restricted ADSs.
| I. | ADS Fees |
The following ADS fees are payable under the terms of the Deposit Agreement:
| Service | Rate | By Whom Paid |
| (1) Issuance of ADSs (e.g., an issuance upon a deposit of Shares, upon a change in the ADS(s)-to-Share(s) ratio, or for any other reason), excluding issuances as a result of distributions described in paragraph (4) below. | Up to U.S. $5.00 per 100 ADSs (or fraction thereof) issued. | Person for whom ADSs are issued. |
| (2) Cancellation of ADSs (e.g., a cancellation of ADSs for Delivery of deposited Shares, upon a change in the ADS(s)-to-Share(s) ratio, or for any other reason). | Up to U.S. $5.00 per 100 ADSs (or fraction thereof) cancelled. | Person for whom ADSs are being cancelled. |
| (3) Distribution of cash dividends or other cash distributions (e.g., upon a sale of rights and other entitlements). | Up to U.S. $5.00 per 100 ADSs (or fraction thereof) held. | Person to whom the distribution is made. |
| (4) Distribution of ADSs pursuant to (i) stock dividends or other free stock distributions, or (ii) an exercise of rights to purchase additional ADSs. | Up to U.S. $5.00 per 100 ADSs (or fraction thereof) held. | Person to whom the distribution is made. |
| (5) Distribution of securities other than ADSs or rights to purchase additional ADSs (e.g., spin-off shares). | Up to U.S. $5.00 per 100 ADSs (or fraction thereof) held. | Person to whom the distribution is made. |
| (6) ADS Services. | Up to U.S. $5.00 per 100 ADSs (or fraction thereof) held on the applicable record date(s) established by the Depositary. | Person holding ADSs on the applicable record date(s) established by the Depositary. |
| (7) Registration of ADS Transfers (e.g., upon a registration of the transfer of registered ownership of ADSs, upon a transfer of ADSs into DTC and vice versa, or for any other reason). | Up to U.S. $5.00 per 100 ADSs (or fraction thereof) transferred. | Person for whom or to whom ADSs are transferred. |
| (8) Conversion of ADSs of one series for ADSs of another series (e.g., upon conversion of Partial Entitlement ADSs for Full Entitlement ADSs, or upon conversion of Restricted ADSs into freely transferable ADSs, and vice versa). | Up to U.S. $5.00 per 100 ADSs (or fraction thereof) converted. | Person for whom ADSs are converted or to whom the converted ADSs are delivered. |
| II. | Charges |
The Company, Holders, Beneficial Owners, persons depositing Shares or withdrawing Deposited Securities in connection with ADS issuances and cancellations, and persons for whom ADSs are issued or cancelled shall be responsible for the following ADS charges under the terms of the Deposit Agreement:
| (i) | taxes (including applicable interest and penalties) and other governmental charges; |
| (ii) | such registration fees as may from time to time be in effect for the registration of Shares or other Deposited Securities on the share register and applicable to transfers of Shares or other Deposited Securities to or from the name of the Custodian, the Depositary or any nominees upon the making of deposits and withdrawals, respectively; |
| (iii) | such cable, telex and facsimile transmission and delivery expenses as are expressly provided in the Deposit Agreement to be at the expense of the person depositing Shares or withdrawing Deposited Property or of the Holders and Beneficial Owners of ADSs; |
| (iv) | in connection with the conversion of Foreign Currency, the fees, expenses, spreads, taxes and other charges of the Depositary and/or conversion service providers (which may be a division, branch or Affiliate of the Depositary). Such fees, expenses, spreads, taxes, and other charges shall be deducted from the Foreign Currency; |
| (v) | any reasonable and customary out-of-pocket expenses incurred in such conversion and/or on behalf of the Holders and Beneficial Owners in complying with currency exchange control or other governmental requirements; and |
| (vi) | the fees, charges, costs and expenses incurred by the Depositary, the Custodian, or any nominee in connection with the ADR program. |
The above fees and charges may at any time and from time to time be changed by agreement between the Company and the Depositary.
B-3
|
●
|
Demand Registration on Form F-1 - each holder is entitled to demand registration on Form F-1, provided that these demand registration rights may only be exercised by holders who hold, in the
aggregate, not less than 30% of the aggregate number of shares held, immediately prior to the completion of our initial public offering, by all holders who are party to the agreement. These demand registration rights may not be
exercised more than twice.
|
| ● |
Demand Registration on Form F-3 - each holder is entitled to demand registration on Form F-3, if we are eligible to register shares on Form F-3, provided that these demand registration
rights may only be exercised by holders who hold, in the aggregate, not less than 20% of the aggregate number of shares held, immediately prior to the completion of our initial public offering, by all holders who are party to the
agreement. These demand registration rights may not be exercised more than twice in any calendar year.
|
| ● |
Piggyback Registration - each holder is entitled to piggyback registration rights, subject, in the case of an underwritten offering, to customary reductions by the underwriter.
|
| ● |
Expenses - We will pay all registration expenses relating to the exercise of the registration rights above, including the reasonable fees and expenses of one legal counsel to the
participating holders up to a maximum of $50,000 in the aggregate.
|
|
●
|
each holder of our ordinary shares is entitled to one vote per ordinary share on all matters to be voted on by shareholders generally;
|
|
●
|
the holders of the ordinary shares shall be entitled to receive notice of, attend, speak and vote at our general meetings; and
|
|
●
|
holders of our ordinary shares are entitled to receive such dividends as are recommended by our directors and declared by our shareholders.
|
| ● |
any resolution put to the vote of a general meeting must be decided exclusively on a poll; on a poll, every shareholder who is present in person or by proxy or corporate representative shall have one vote for
each share of which they are the holder. A shareholder entitled to more than one vote need not, if they vote, use all their votes or cast all the votes in the same way; and
|
| ● |
if two or more persons are joint holders of a share, then in voting on any question the vote of the senior who tenders a vote, whether in person or by proxy, shall be accepted to the exclusion of the votes of the other joint holders. For
this purpose seniority shall be determined by the order in which the names of the holders stand in the share register.
|
| ● |
it is for a share which is fully paid up;
|
| ● |
it is for a share upon which we have no lien;
|
| ● |
it is only for one class of share;
|
| ● |
it is in favor of a single transferee or no more than four joint transferees;
|
| ● |
it is duly stamped or is duly certificated or otherwise shown to the satisfaction of the board to be exempt from stamp duty (if this is required); and
|
| ● |
it is delivered for registration to our registered office (or such other place as the board may determine), accompanied (except in the case of a transfer by a person to whom we are not required by law to issue a
certificate and to whom a certificate has not been issued or in the case of a renunciation) by the certificate for the shares to which it relates and such other evidence as the board may reasonably require to prove the title of the
transferor (or person renouncing) and the due execution of the transfer or renunciation by him or, if the transfer or renunciation is executed by some other person on his behalf, the authority of that person to do so.
|
The board of directors may decline to register a transfer of uncertificated shares in any circumstances that are allowed or required by the Uncertificated Securities Regulations 2001 and the requirements of its relevant system.
| ● |
the quorum for such class meeting shall be two holders in person or by proxy representing not less than one-third in nominal value of the issued shares of the class (excluding any shares held in treasury); and
|
| ● |
if at any adjourned meeting of such holders a quorum is not present at the meeting, one holder of shares of the class present in person or by proxy at an adjourned meeting constitutes a quorum.
|
| ● |
the giving of any guarantee, security or indemnity in respect of money lent or obligations incurred by him or by any other person at the request of or for the benefit of our company or any of our subsidiary undertakings;
|
| ● |
the giving of any guarantee, security or indemnity in respect of a debt or obligation of our company or any of our subsidiary undertakings for which he himself has assumed responsibility in whole or in part under a guarantee or indemnity
or by the giving of security;
|
| ● |
any proposal or contract relating to an offer of securities of or by our company or any of our subsidiary undertakings in which offer he is or may be entitled to participate as a holder of securities or in the underwriting or
sub-underwriting of which he is to participate;
|
| ● |
any arrangement involving any other company if the director (together with any person connected with him) has an interest of any kind in that company (including an interest by holding any position in that
company or by being a member of that company), unless he is to his knowledge (either directly or indirectly) the holder of or beneficially interested in one per cent or more of any class of the equity share capital of that company
(calculated exclusive of any shares of that class in that company held as treasury shares) or of the voting rights available to members of that company;
|
| ● |
any arrangement for the benefit of employees of our company or any of our subsidiary undertakings which only gives him benefits which are also generally given to employees to whom the arrangement relates;
|
| ● |
any contract relating to insurance which our company is to buy or renew for the benefit of the directors or a group of people which includes directors; and
|
| ● |
a contract relating to a pension, superannuation or similar scheme or a retirement, death, disability benefits scheme or employees’ share scheme which gives the director benefits which are also generally given to the employees to whom
the scheme relates.
|
|
|
England and Wales
|
|
Delaware
|
|
|
Number of Directors
|
|
Under the Companies Act, a public limited company must have at least two directors and the number of directors may be fixed by or in the manner provided in a company’s articles of association.
|
|
Under Delaware law, a corporation must have at least one director and the number of directors shall be fixed by or in the manner provided in the bylaws.
|
|
|
|
|||
|
Removal of Directors
|
|
Under the Companies Act, shareholders may remove a director without cause by an ordinary resolution (which is passed by a simple majority of those voting in person or by proxy at a general meeting) irrespective of any provisions of any
service contract the director has with the Company, provided 28 clear days’ notice of the resolution has been given to the Company and its shareholders. On receipt of notice of an intended resolution to remove a director, the Company must
forthwith send a copy of the notice to the director concerned. Certain other procedural requirements under the. Companies Act must also be followed such as allowing the director to make representations against his or her removal either at
the meeting or in writing.
|
|
Under Delaware law, any director or the entire board of directors may be removed, with or without cause, by the holders of a majority of the shares then entitled to vote at an election of directors, except (a) unless the certificate of
incorporation provides otherwise, in the case of a corporation whose board of directors is classified, shareholders may effect such removal only for cause, or (b) in the case of a corporation having cumulative voting, if less than the
entire board of directors is to be removed, no director may be removed without cause if the votes cast against his removal would be sufficient to elect him if then cumulatively voted at an election of the entire board of directors, or, if
there are classes of directors, at an election of the class of directors of which he is a part.
|
|
|
|
|||
|
Vacancies on the Board of Directors
|
|
Under the laws of England and Wales, the procedure by which directors, other than a company’s initial directors, are appointed is generally set out in a company’s articles of association, provided that where two or more persons are
appointed as directors of a public limited company by resolution of the shareholders, resolutions appointing each director must be voted on individually.
|
|
Under Delaware law, vacancies and newly created directorships may be filled by a majority of the directors then in office (even though less than a quorum) or by a sole remaining director unless (a) otherwise provided in the certificate
of incorporation or by-laws of the corporation or (b) the certificate of incorporation directs that a particular class of stock is to elect such director, in which case a majority of the other directors elected by such class, or a sole
remaining director elected by such class, will fill such vacancy.
|
|
|
England and Wales
|
|
Delaware
|
|
|
|
|
|||
|
Annual General Meeting
|
|
Under the Companies Act, a public limited company must hold an annual general meeting in each six-month period following its annual accounting reference date.
|
|
Under Delaware law, the annual meeting of stockholders shall be held at such place, on such date and at such time as may be designated from time to time by the board of directors or as provided in the certificate of incorporation or by
the bylaws.
|
|
|
|
|||
|
General Meeting
|
|
Under the Companies Act, a general meeting of the shareholders of a public limited company may be called by the directors.
Shareholders holding at least 5% of the paid-up capital of the Company carrying voting rights at general meetings (excluding any paid up capital held as treasury shares) can require the directors to call a general meeting and, if the
directors fail to do so within a certain period, may themselves (or any of them representing more than one half of the total voting rights of all of them) convene a general meeting.
|
|
Under Delaware law, special meetings of the stockholders may be called by the board of directors or by such person or persons as may be authorized by the certificate of incorporation or by the bylaws.
|
|
|
|
|||
|
Notice of General Meetings
|
|
Subject to a company’s articles of association providing for a longer period, under the Companies Act, 21 clear days’ notice must be given for an annual general meeting and any resolutions to be proposed at the meeting. Subject to a
company’s articles of association providing for a longer period, at least 14 clear days’ notice is required for any other general meeting. In addition, certain matters, such as the removal of directors or auditors, require special notice,
which is 28 clear days’ notice. The shareholders of a company may in all cases consent to a shorter notice period, the proportion of shareholders’ consent required being 100% of those entitled to attend and vote in the case of an annual
general meeting and, in the case of any other general meeting, a majority in number of the members having a right to attend and vote at the meeting, being a majority who together hold not less than 95% in nominal value of the shares giving
a right to attend and vote at the meeting.
|
|
Under Delaware law, unless otherwise provided in the certificate of incorporation or bylaws, written notice of any meeting of the stockholders must be given to each stockholder entitled to vote at the meeting not less than 10 nor more
than 60 days before the date of the meeting and shall specify the place, date, hour, and purpose or purposes of the meeting.
|
|
|
England and Wales
|
|
Delaware
|
|
|
|
|
|||
|
Quorum
|
|
Subject to the provisions of a company’s articles of association, the Companies Act provides that two shareholders present at a meeting (in person, by proxy or authorized representative under the Companies Act) shall constitute a quorum
for companies with more than one member.
|
|
The certificate of incorporation or bylaws may specify the number of shares, the holders of which shall be present or represented by proxy at any meeting in order to constitute a quorum, but in no event shall a quorum consist of less
than one third of the shares entitled to vote at the meeting. In the absence of such specification in the certificate of incorporation or bylaws, a majority of the shares entitled to vote, present in person or represented by proxy, shall
constitute a quorum at a meeting of stockholders.
|
|
|
|
|||
|
Proxy
|
|
Under the Companies Act, at any meeting of shareholders, a shareholder may designate another person to attend, speak and vote at the meeting on their behalf by proxy.
|
|
Under Delaware law, at any meeting of stockholders, a stockholder may designate another person to act for such stockholder by proxy, but no such proxy shall be voted or acted upon after three years from its date, unless the proxy
provides for a longer period. A director of a Delaware corporation may not issue a proxy representing the director’s voting rights as a director.
|
|
|
|
|||
|
Preemptive Rights
|
|
Under the Companies Act, “equity securities,” being (1) shares in the Company other than shares that, with respect to dividends and capital, carry a right to participate only up to a specified amount in a distribution, referred to as
“ordinary shares,” or (2) rights to subscribe for, or to convert securities into, ordinary shares, proposed to be allotted for cash must be offered first to the existing equity shareholders in the Company in proportion to the respective
nominal value of their holdings, unless an exception applies or a special resolution to the contrary has been passed by shareholders in a general meeting or the articles of association provide otherwise in each case in accordance with the
provisions of the Companies Act.
|
|
Under Delaware law, shareholders have no preemptive rights to subscribe to additional issues of stock or to any security convertible into such stock unless, and except to the extent that, such rights are expressly provided for in the
certificate of incorporation.
|
|
|
|
|||
|
Authority to Allot
|
|
Under the Companies Act, the directors of a company must not allot shares or grant of rights to subscribe for or to convert any security into shares unless an exception applies or an ordinary resolution to the contrary has been passed by
shareholders in a general meeting or the articles of association provide otherwise in each case in accordance with the provisions of the Companies Act.
|
|
Under Delaware law, if the corporation’s charter or certificate of incorporation so provides, the board of directors has the power to authorize the issuance of stock. It may authorize capital stock to be issued for consideration
consisting of cash, any tangible or intangible property or any benefit to the corporation or any combination thereof. It may determine the amount of such consideration by approving a formula. In the absence of actual fraud in the
transaction, the judgment of the directors as to the value of such consideration is conclusive.
|
|
|
England and Wales
|
|
Delaware
|
|
|
|
|
|||
|
Liability of Directors and Officers
|
|
Under the Companies Act, any provision, whether contained in a company’s articles of association or any contract or otherwise, that purports to exempt a director of a company, to any extent, from any liability that would otherwise attach
to him in connection with any negligence, default, breach of duty or breach of trust in relation to the Company is void.
Any provision by which a company directly or indirectly provides an indemnity, to any extent, for a director of the Company or of an associated company against any liability attaching to him in connection with any negligence, default,
breach of duty or breach of trust in relation to the Company of which he is a director is also void except as permitted by the Companies Act, which provides exceptions for the Company to (a) purchase and maintain insurance against such
liability; (b) provide a “qualifying third party indemnity” (being an indemnity against liability incurred by the director to a person other than the Company or an associated company or criminal proceedings in which he is convicted); and
(c) provide a “qualifying pension scheme indemnity” (being an indemnity against liability incurred in connection with our activities as trustee of an occupational pension plan).
|
|
Under Delaware law, a corporation’s certificate of incorporation may include a provision eliminating or limiting the personal liability of a director to the corporation and its stockholders for damages arising from a breach of fiduciary
duty as a director. However, no provision can limit the liability of a director for:
● any breach of the director’s duty of loyalty to the corporation or its stockholders;
● acts or omissions not in good faith or that involve intentional misconduct or a knowing violation of law;
● intentional or negligent payment of unlawful dividends or stock purchases or redemptions; or
● any transaction from which the director derives an improper personal benefit.
|
|
|
|
|||
|
Voting Rights
|
|
For a company incorporated under the laws of England and Wales, it is usual for the articles of association to provide that, unless a poll is demanded by the shareholders of a company or is required by the chairman of the meeting or our
articles of association, shareholders shall vote on all resolutions on a show of hands. Under the Companies Act, a poll may be demanded by (a) not fewer than five shareholders having the right to vote on the resolution; (b) any
shareholder(s) representing not less than 10% of the total voting rights of all the shareholders having the right to vote on the resolution (excluding any voting rights attaching to treasury shares); or (c) any shareholder(s) holding shares
in the Company conferring a right to vote on the resolution (excluding any voting rights attaching to treasury shares) being shares on which an aggregate sum has been paid up equal to not less than 10% of the total sum paid up on all the
shares conferring that right. A company’s articles of association may provide more extensive rights for shareholders to call a poll.
Under the laws of England and Wales, an ordinary resolution is passed on a show of hands if it is approved by a simple majority (more than 50%) of the votes cast by shareholders present (in person or by proxy) and entitled to vote. If
a poll is demanded, an ordinary resolution is passed if it is approved by holders representing a simple majority of the total voting rights of shareholders present, in person or by proxy, who, being entitled to vote, vote on the
resolution. Special resolutions require the affirmative vote of not less than 75% of the votes cast by shareholders present, in person or by proxy, at the meeting. If a poll is demanded, a special resolution is passed if it is approved by
holders representing not less than 75% of the total voting rights of shareholders in person or by proxy who, being entitled to vote, vote on the resolution.
|
|
Delaware law provides that, unless otherwise provided in the certificate of incorporation, each stockholder is entitled to one vote for each share of capital stock held by such stockholder.
|
|
|
England and Wales
|
|
Delaware
|
|
|
|
|
|||
|
Shareholder Vote on Certain Transactions
|
|
The Companies Act provides for schemes of arrangement, which are arrangements or compromises between a company and any class of shareholders or creditors and used in certain types of reconstructions, amalgamations, capital
reorganizations, or takeovers. These arrangements require:
● the approval at a shareholders’ or creditors’ meeting convened by order of the court, of a majority in number of shareholders or
creditors or a class thereof representing 75% in value of the capital held by, or debt owed to, the class of shareholders or creditors, or class thereof present and voting, either in person or by proxy; and
● the approval of the court.
|
|
Generally, under Delaware law, unless the certificate of incorporation provides for the vote of a larger portion of the stock, completion of a merger, consolidation, sale, lease or exchange of all or substantially all of a corporation’s
assets or dissolution requires:
● the approval of the board of directors; and
● approval by the vote of the holders of a majority of the outstanding stock or, if the certificate of incorporation provides for more
or less than one vote per share, a majority of the votes of the outstanding stock of a corporation entitled to vote on the matter.
|
|
Standard of Conduct for Directors
|
Under the laws of England and Wales, a director owes various statutory and fiduciary duties to the Company, including:
● to act in the way he considers, in good faith, would be most likely to promote the success of the Company for the benefit of its
members as a whole, and in doing so have regard (amongst other matters) to: (i) the likely consequences of any decision in the long-term, (ii) the interests of the company’s employees, (iii) the need to foster the company’s business
relationships with suppliers, customers and others, (iv) the impact of the company’s operations on the community and the environment, (v) the desirability to maintain a reputation for high standards of business conduct, and (vi) the need to
act fairly as between members of the company;
● to avoid a situation in which he has, or can have, a direct or indirect interest that conflicts, or possibly conflicts, with the
interests of the Company;
● to act in accordance with our constitution and only exercise his powers for the purposes for which they are conferred;
● to exercise independent judgment;
● to exercise reasonable care, skill, and diligence;
● not to accept benefits from a third party conferred by reason of his being a director or doing, or not doing, anything as a director;
and
● a duty to declare any interest that he has, whether directly or indirectly, in a proposed or existing transaction or arrangement with
the Company.
|
Delaware law does not contain specific provisions setting forth the standard of conduct of a director. The scope of the fiduciary duties of directors is generally determined by the courts of the State of Delaware. In general, directors
have a duty to act without self-interest, on a well-informed basis and in a manner they reasonably believe to be in the best interest of the stockholders.
Directors of a Delaware corporation owe fiduciary duties of care and loyalty to the corporation and to its shareholders. The duty of care generally requires that a director act in good faith, with the care that an ordinarily prudent
person would exercise under similar circumstances. Under this duty, a director must inform himself of all material information reasonably available regarding a significant transaction. The duty of loyalty requires that a director act in a
manner he reasonably believes to be in the best interests of the corporation. He must not use his corporate position for personal gain or advantage. In general, but subject to certain exceptions, actions of a director are presumed to have
been made on an informed basis, in good faith and in the honest belief that the action taken was in the best interests of the corporation. However, this presumption may be rebutted by evidence of a breach of one of the fiduciary duties.
Delaware courts have also imposed a heightened standard of conduct upon directors of a Delaware corporation who take any action designed to defeat a threatened change in control of the corporation.
In addition, under Delaware law, when the board of directors of a Delaware corporation approves the sale or break-up of a corporation, the board of directors may, in certain circumstances, have a duty to obtain the highest value
reasonably available to the shareholders.
|
|
|
England and Wales
|
|
Delaware
|
|
|
Shareholder Litigation
|
|
Under the laws of England and Wales, generally, the Company, rather than its shareholders, is the proper claimant in an action in respect of a wrong done to the Company or where there is an irregularity in the Company’s internal
management. Notwithstanding this general position, the Companies Act provides that (1) a court may allow a shareholder to bring a derivative claim (that is, an action in respect of and on behalf of the Company) in respect of a cause of
action arising from a director’s negligence, default, breach of duty or breach of trust and (2) a shareholder may bring a claim for a court order where our affairs have been or are being conducted in a manner that is unfairly prejudicial to
some of its shareholders.
|
|
Under Delaware law, a stockholder may initiate a derivative action to enforce a right of a corporation if the corporation fails to enforce the right itself. The complaint must:
● state that the plaintiff was a stockholder at the time of the transaction of which the plaintiff complains or that the plaintiffs
shares thereafter devolved on the plaintiff by operation of law; and
● allege with particularity the efforts made by the plaintiff to obtain the action the plaintiff desires from the directors and the
reasons for the plaintiff’s failure to obtain the action; or
● state the reasons for not making the effort.
Additionally, the plaintiff must remain a stockholder through the duration of the derivative suit. The action will not be dismissed or compromised without the approval of the Delaware Court of Chancery.
|
| ● |
we do not timely request that the rights be distributed to the holders or we request that the rights not be distributed to the holders; or
|
| ● |
we fail to deliver satisfactory documents to the depositary; or
|
| ● |
it is not reasonably practicable to distribute the rights.
|
| ● |
we do not request that the property be distributed to the holders or if we ask that the property not be distributed to the holders;
|
| ● |
we do not deliver satisfactory documents to the depositary; or
|
| ● |
the depositary determines that all or a portion of the distribution is not reasonably practicable.
|
| ● |
ensure that the surrendered ADR is properly endorsed or otherwise in proper form for transfer;
|
| ● |
provide such proof of identity and genuineness of signatures, and of such other matters contemplated in the Deposit Agreement, as the depositary deems appropriate;
|
| ● |
comply with applicable laws and regulations, including regulations imposed by us and the depositary consistent with the Deposit Agreement, the ADR and applicable law;
|
| ● |
provide any transfer stamps required by the State of New York or the United States; and
|
| ● |
pay all applicable fees, charges, expenses, taxes and other government charges payable by ADR holders pursuant to the terms of the Deposit Agreement, upon the transfer of ADRs.
|
| ● |
temporary delays that may arise because (1) the transfer books for the ordinary shares or ADSs are closed, or (2) ordinary shares are immobilized on account of a shareholders’ meeting or a payment of dividends;
|
| ● |
obligations to pay fees, taxes and similar charges; or
|
| ● |
restrictions imposed because of laws or regulations applicable to ADSs or the withdrawal of securities on deposit.
|
| ● |
In the event of voting by show of hands, the depositary will vote (or cause the custodian to vote) all ordinary shares held on deposit at that time in accordance with the voting instructions
received from a majority of holders of ADSs who provide timely voting instructions.
|
| ● |
In the event of voting by poll, the depositary will vote (or cause the custodian to vote) the ordinary shares held on deposit in accordance with the voting instructions received from the holders
of ADSs.
|
|
Service
|
|
Fee
|
|
Issuance of ADSs (e.g., an issuance of ADS upon a deposit of ordinary shares or upon a change in the ADS(s)-to-ordinary shares ratio, or for any other reason), excluding ADS issuances as a result
of distributions of ordinary shares
|
|
Up to $0.05 per ADS issued
|
|
|
||
|
Cancellation of ADSs (e.g., a cancellation of ADSs for delivery of deposited property or upon a change in the ADS(s)-to-ordinary shares ratio, or for any other reason)
|
|
Up to $0.05 per ADS cancelled
|
|
|
||
|
Distribution of cash dividends or other cash distributions (e.g., upon a sale of rights and other entitlements)
|
|
Up to $0.05 per ADS held
|
|
|
||
|
Distribution of ADSs pursuant to (i) share dividends or other distributions, or (ii) exercise of rights to purchase additional ADSs
|
|
Up to $0.05 per ADS held
|
|
|
||
|
Distribution of securities other than ADSs or rights to purchase additional ADSs (e.g., upon a spin-off)
|
|
Up to $0.05 per ADS held
|
|
|
||
|
ADS services
|
|
Up to $0.05 per ADS held on the applicable record date(s) established by the depositary
|
| ● |
taxes (including applicable interest and penalties) and other governmental charges;
|
| ● |
the registration fees as may from time to time be in effect for the registration of ordinary shares on the share register and applicable to transfers of ordinary shares to or from the name of the custodian, the depositary or any nominees
upon the making of deposits and withdrawals, respectively;
|
| ● |
certain cable, telex and facsimile transmission and delivery expenses;
|
| ● |
the expenses and charges incurred by the depositary in the conversion of foreign currency;
|
| ● |
the fees and expenses incurred by the depositary in connection with compliance with exchange control regulations and other regulatory requirements applicable to ordinary shares, ADSs and ADRs; and
|
| ● |
the fees and expenses incurred by the depositary, the custodian, or any nominee in connection with the servicing or delivery of deposited property.
|
| ● |
We and the depositary are obligated only to take the actions specifically stated in the Deposit Agreement without negligence or bad faith.
|
| ● |
The depositary disclaims any liability for any failure to carry out voting instructions, for any manner in which a vote is cast or for the effect of any vote, provided it acts in good faith and in accordance with the terms of the Deposit
Agreement.
|
| ● |
The depositary disclaims any liability for any failure to accurately determine the lawfulness or practicality of any action, for the content of any document forwarded to holders on our behalf or for the accuracy of any translation of
such a document, for the investment risks associated with investing in ordinary shares, for the validity or worth of the ordinary shares, for any tax consequences that result from the ownership of ADSs or other deposited property, for the
credit-worthiness of any third party, for allowing any rights to lapse under the terms of the Deposit Agreement, for the timeliness of any of our notices or for our failure to give notice or for any act or omission of or information
provided by DTC or any DTC participant.
|
| ● |
The depositary shall not be liable for acts or omissions of any successor depositary in connection with any matter arising wholly after the resignation or removal of the depositary.
|
| ● |
We and the depositary will not be obligated to perform any act that is inconsistent with the terms of the Deposit Agreement.
|
| ● |
We and the depositary disclaim any liability if we or the depositary are prevented or forbidden from or subject to any civil or criminal penalty or restraint on account of, or delayed in, doing or performing any act or thing required by
the terms of the Deposit Agreement, by reason of any provision, present or future of any law or regulation, including regulations of any stock exchange or by reason of present or future provisions of our articles of association, or any
provision of or governing the securities on deposit, or by reason of any act of God or war or other circumstances beyond our or the depositary’s control.
|
| ● |
We and the depositary disclaim any liability by reason of any exercise of, or failure to exercise, any discretion provided for in the Deposit Agreement or in our articles of association or in any provisions of or governing the securities
on deposit.
|
| ● |
We and the depositary further disclaim any liability for any action or inaction in reliance on the advice or information received from legal counsel, accountants, any person presenting ordinary shares for deposit, any holder of ADSs or
authorized representatives thereof, or any other person believed by either of us in good faith to be competent to give such advice or information.
|
| ● |
We and the depositary also disclaim liability for the inability by any ADS holder or beneficiary owner to benefit from any distribution, offering, right or other benefit that is made available to holders of ordinary shares but is not,
under the terms of the Deposit Agreement, made available to any applicable holder.
|
| ● |
We and the depositary may rely without any liability upon any written notice, request or other document believed to be genuine and to have been signed or presented by the proper parties.
|
| ● |
We and the depositary also disclaim liability for any consequential or punitive damages for any breach of the terms of the Deposit Agreement.
|
| ● |
We and the depositary disclaim liability arising out of losses, liabilities, taxes, charges or expenses resulting from the manner in which a holder or beneficial owner of ADSs holds ADSs, including resulting from holding ADSs through a
brokerage account.
|
| ● |
No disclaimer of any Securities Act liability is intended by any provision of the Deposit Agreement.
|
| ● |
Nothing in the Deposit Agreement gives rise to a partnership or joint venture, or establishes a fiduciary relationship, among us, the depositary and any ADS holder.
|
| ● |
Nothing in the Deposit Agreement precludes Citibank (or its affiliates) from engaging in transactions in which parties adverse to us or the ADS owners have interests, and nothing in the Deposit Agreement obligates Citibank to disclose
those transactions, or any information obtained in the course of those transactions, to us or to the ADS owners, or to account for any payment received as part of those transactions.
|
| ● |
Convert the foreign currency to the extent practical and lawful and distribute the U.S. dollars to the ADS holders for whom the conversion and distribution is lawful and practical.
|
| ● |
Distribute the foreign currency to ADS holders for whom the distribution is lawful and practical.
|
| ● |
Hold the foreign currency (without liability for interest) for the applicable ADS holders.
|
Exhibit 4.20
IMMUNOCORE HOLDINGS PLC
2021 Equity Incentive Plan
With
Non-Employee Sub-Plan
Adopted by the Board of Directors: 28 January 2021
Approved by the Shareholders: 3 February 2021
Share Reserve Approved by the Pricing Committee of the Board of Directors: 4 February 2021
IPO Date: 4 February 2021
Table of contents
| Page | |
| 1. PURPOSE | 1 |
| 2. ELIGIBILITY | 1 |
| 3. ADMINISTRATION AND DELEGATION. | 1 |
| 4. SHARES AVAILABLE FOR AWARDS. | 1 |
| 5. OPTIONS AND SHARE APPRECIATION RIGHTS. | 3 |
| 6. RESTRICTED SHARES; RESTRICTED SHARE UNITS | 5 |
| 7. OTHER SHARE BASED AWARDS | 6 |
| 8. ADJUSTMENTS FOR CHANGES IN SHARES AND CERTAIN OTHER EVENTS | 6 |
| 9. GENERAL PROVISIONS APPLICABLE TO AWARDS | 8 |
| 10. MISCELLANEOUS | 10 |
| 11. Covenants of the Company. | 15 |
| 12. DEFINITIONS. | 15 |
1. PURPOSE
The Plan’s purpose is to enhance the Company’s ability to attract, retain and motivate persons who make (or are expected to make) important contributions to the Company by providing these individuals with equity ownership opportunities. Capitalized terms used in the Plan are defined in Section 12.
2. ELIGIBILITY
Service Providers are eligible to be granted Awards under the Plan, subject to the limitations described herein.
3. ADMINISTRATION AND DELEGATION.
(a) Administration. The Plan is administered by the Administrator. The Administrator has authority to (i) determine which Service Providers receive Awards, (ii) grant Awards, (iii) set Award terms and conditions, and (iv) designate whether such Awards will cover Ordinary Shares or ADSs, in each case subject to the conditions and limitations in the Plan. The Administrator also has the authority to take all actions and make all determinations under the Plan, to approve the forms of Award Agreements for use under the Plan, to interpret the Plan and the terms of Awards and to adopt, amend and repeal Plan administrative rules, guidelines and practices as it deems advisable. The Administrator may correct defects and ambiguities, supply omissions and reconcile inconsistencies in the Plan or any Award as it deems necessary or appropriate to administer the Plan and any Awards. The Administrator’s determinations under the Plan are in its sole discretion and will be final and binding on all persons having or claiming any interest in the Plan or any Award.
(b) Appointment of Committees. To the extent Applicable Laws permit, the Board may delegate any or all of its powers under the Plan to one or more Committees or officers of the Company or any of its Subsidiaries. The Board may abolish any Committee or re-vest in itself any previously delegated authority at any time.
4. SHARES AVAILABLE FOR AWARDS.
(a) Number of Shares. Subject to adjustment under Section 8 and the terms of this Section 4, Awards may be made under the Plan (taking account of Awards granted under the Non-Employee Sub-Plan) in an aggregate amount up to 5,992,994 Ordinary Shares plus any Ordinary Shares that become available under the Plan pursuant to Section 4(c)(ii) below (in each case including as part of the process for the issue of new ADSs) (the “Share Reserve”). In addition, the Share Reserve will automatically increase on January 1st of each year commencing on January 1, 2022 and ending on (and including) January 1, 2031, in an amount equal to 5% of the total number of Ordinary Shares outstanding on December 31st of the preceding calendar year. Notwithstanding the foregoing, the Board may act prior to January 1st of a given year to provide that there will be no January 1st increase in the Share Reserve for such year or that the increase in the Share Reserve for such year will be a lesser (but not a greater) number of Shares than would otherwise occur pursuant to the preceding sentence.
(b) Limit Applies to Shares Issued Pursuant to Awards. For clarity, the Share Reserve is a limit on the number of Shares that may be issued pursuant to Awards that were granted under this Plan and does not limit the granting of Awards, except that the Company will keep available at all times the number of Shares reasonably required to satisfy its obligations to issue shares pursuant to such Awards. Shares may be issued in connection with a merger or acquisition as permitted by, as applicable, Nasdaq Listing Rule 5635(c), NYSE Listed Company Manual Section 303A.08, NYSE American Company Guide Section 711 or other applicable rule, and such issuance will not reduce the number of Shares available for issuance under the Plan, as further described under Section 4(e).
(c) Share Recycling.
(i) If all or any part of an Award or Awards granted under the Plan (including the Non-Employee Sub-Plan) expires, lapses or is terminated, exchanged for cash, surrendered, repurchased or cancelled without having been fully exercised, or is withheld to satisfy a tax withholding obligation in connection with an Award or to satisfy a purchase or exercise price of an Award, the unused Shares covered by the Award or Awards granted under the Plan (including the Non-Employee Sub-Plan) will, as applicable, become or again be available for Awards granted under the Plan (including the Non-Employee Sub-Plan).
(ii) If all or any part of an option or options to acquire unissued Shares that was granted under the Prior Plans and which is subsisting as of the Effective Date expires, lapses or is terminated, exchanged for cash, surrendered, repurchased or cancelled without having been fully exercised, or is withheld to satisfy a tax withholding obligation in connection with an option or to satisfy a purchase or exercise price of an option, in each case on or after the Effective Date, the unused Shares covered by such option or options under the Prior Plan shall increase the Share Reserve and shall become available for Awards granted under the Plan (including the Non-Employee Sub-Plan) subject to a maximum of 4,551,360 Ordinary Shares (including as part of the process for the issue of new ADSs).
(d) ISO Limitations. Subject to adjustment under Section 8 and to the overall Share Reserve, no more than 10,544,354 Ordinary Shares (including as part of the process for the issue of new ADSs) may be issued pursuant to the exercise of ISOs.
(e) Substitute Awards. In connection with an entity’s merger or consolidation with the Company or the Company’s acquisition of an entity’s property or stock, the Administrator may grant Awards in substitution for any options or other equity or equity-based awards granted before such merger or consolidation by such entity or its affiliate. Substitute Awards may be granted on such terms as the Administrator deems appropriate, notwithstanding limitations on Awards in the Plan. Subject to Applicable Laws, Substitute Awards will not count against the Share Reserve (nor shall Shares subject to a Substitute Award be added to the Shares available for Awards under the Plan as provided above), except that Shares acquired by exercise of substitute ISOs will count against the maximum number of Shares that may be issued pursuant to the exercise of ISOs under the Plan. Additionally, in the event that a company acquired by the Company or any Subsidiary or with which the Company or any Subsidiary combines has shares available under a pre-existing plan not adopted in contemplation of such acquisition or combination, then, subject to Applicable Laws, shares available for grant pursuant to the terms of such pre-existing plan (as adjusted, to the extent appropriate, using the exchange ratio or other adjustment or valuation ratio or formula used in such acquisition or combination to determine the consideration payable to the holders of ordinary shares or common stock (as applicable) of the entities party to such acquisition or combination) may be used for Awards under the Plan and shall not reduce the Shares authorized for grant under the Plan (and Shares subject to such Awards shall not be added to the Shares available for Awards under the Plan as provided above); provided that Awards using such available shares shall not be made after the date awards or grants could have been made under the terms of the pre-existing plan, absent the acquisition or combination, and shall only be made to individuals who were not Employees or Directors prior to such acquisition or combination.
(f) Date of Grant. Unless otherwise determined by the Administrator, the date of grant of an Award shall be the date of the Administrator’s approval of that Award.
(g) Deed Poll. The Administrator may grant Awards by entering into a deed poll and, as soon as practicable after the Company has executed the deed poll, the Administrator shall enter into an Award Agreement.
(h) Prior Plans. Upon the Effective Date, no further new awards may be granted over Shares under the Prior Plans.
5. OPTIONS AND SHARE APPRECIATION RIGHTS.
(a) General. The Administrator may grant Options or Share Appreciation Rights to Service Providers subject to the limitations in the Plan, including any limitations in the Plan that apply to ISOs. The Administrator will determine the number of Shares covered by each Option and Share Appreciation Right, the exercise price of each Option and Share Appreciation Right and the conditions and limitations applicable to the exercise of each Option and Share Appreciation Right. Each Option will be designated in writing as an ISO or Non-Qualified Option at the time of grant; provided, however, that if an Option is not so designated, then such Option will be a Non-Qualified Option, and the Shares purchased upon exercise of each type of Option will be separately accounted for. A Share Appreciation Right will entitle the Participant (or other person entitled to exercise the Share Appreciation Right) to receive from the Company upon exercise of the exercisable portion of the Share Appreciation Right an amount determined by multiplying the excess, if any, of the Fair Market Value of one Share on the date of exercise over the exercise price per Share of the Share Appreciation Right by the number of Shares with respect to which the Share Appreciation Right is exercised, subject to any limitations of the Plan or that the Administrator may impose and payable in cash, Shares valued at Fair Market Value or a combination of the two as the Administrator may determine or provide in the Award Agreement. A Participant will have no rights of a shareholder with respect to Shares subject to any Option or Share Appreciation Right unless and until any Shares are delivered in settlement of the Option or Share Appreciation Right.
(b) Exercise Price. The Administrator will establish each Option’s and Share Appreciation Right’s exercise price and specify the exercise price in the Award Agreement. Subject to Section 10(g), the exercise price will not be less than 100% of the Fair Market Value on the grant date of the Option or Share Appreciation Right. Notwithstanding the foregoing, an Option or Share Appreciation Right may be granted with an exercise price lower than 100% of the Fair Market Value on the date of grant of such Award if such Award is granted pursuant to an assumption of or substitution for another option or share appreciation right pursuant to Section 4(e) and, in respect of Participants who are subject to tax in the United States, in a manner consistent with the provisions of Sections 409A and, if applicable, 424(a) of the Code.
(c) Duration. Each Option or Share Appreciation Right will vest and be exercisable at such times and as specified in the Award Agreement, provided that the term of an Option or Share Appreciation Right will not exceed ten years, subject to Section 10(g). Notwithstanding the foregoing and unless determined otherwise by the Company, in the event that on the last business day of the term of an Option or Share Appreciation Right (other than an ISO) (i) the exercise of the Option or Share Appreciation Right is prohibited by Applicable Laws, as determined by the Company, or (ii) Shares may not be purchased or sold by the applicable Participant due to any Company insider trading, window period and/or dealing policy (including blackout periods), the term of the Option or Share Appreciation Right shall be extended until the date that is thirty (30) days after the end of the legal prohibition, black-out period, as determined by the Company; provided, however, in no event shall the extension last beyond the ten year term of the applicable Option or Share Appreciation Right. Notwithstanding the foregoing, if the Participant, prior to the end of the term of an Option or Share Appreciation Right, violates the non-competition, non-solicitation, confidentiality or other similar restrictive covenant provisions of any employment contract, confidentiality and nondisclosure agreement or other agreement between the Participant and the Company or any of its Subsidiaries, the right of the Participant and the Participant’s transferees to exercise any Option or Share Appreciation Right issued to the Participant shall terminate immediately upon such violation, unless the Company otherwise determines. In addition, if, prior to the end of the term of an Option or Share Appreciation Right, the Participant is given notice by the Company or any of its Subsidiaries of the Participant’s Termination of Service by the Company or any of its Subsidiaries for Cause, and the effective date of such Termination of Service is subsequent to the date of the delivery of such notice, the right of the Participant and the Participant’s transferees to exercise any Option or Share Appreciation Right issued to the Participant shall be suspended from the time of the delivery of such notice until the earlier of (i) such time as it is determined or otherwise agreed that the Participant’s service as a Service Provider will not be terminated for Cause as provided in such notice or (ii) the effective date of the Participant’s Termination of Service by the Company or any of its Subsidiaries for Cause (in which case the right of the Participant and the Participant’s transferees to exercise any Option or Share Appreciation Right issued to the Participant will terminate immediately upon the effective date of such Termination of Service).
(d) Exercise. Options and Share Appreciation Rights may be exercised by delivering to the Company a written notice of exercise, in a form the Administrator approves (which may be electronic), signed by the person authorized to exercise the Option or Share Appreciation Right, together with, as applicable, payment in full (i) as specified in Section 5(e) for the number of Shares for which the Award is exercised and (ii) as specified in Section 9(e) for any applicable taxes. Unless the Administrator otherwise determines, an Option or Share Appreciation Right may not be exercised for a fraction of a Share.
(e) Payment Upon Exercise. Subject to any Company insider trading, window period and/or dealing policy (including blackout periods) and Applicable Laws, the exercise price of an Option must be paid by:
(i) cash, wire transfer of immediately available funds or by check payable to the order of the Company, provided that the Company may limit the use of one of the foregoing payment forms if one or more of the payment forms below is permitted;
(ii) if there is a public market for Shares at the time of exercise, unless the Company otherwise determines, (A) delivery (including telephonically to the extent permitted by the Company) of an irrevocable and unconditional undertaking by a broker acceptable to the Company to deliver promptly to the Company sufficient funds to pay the exercise price, or (B) the Participant’s delivery to the Company of a copy of irrevocable and unconditional instructions to a broker acceptable to the Company to deliver promptly to the Company cash or a check sufficient to pay the exercise price; provided that such amount is paid to the Company at such time as may be required by the Administrator;
(iii) to the extent permitted by the Administrator at the time of exercise, delivery (either by actual delivery or attestation) of Shares owned by the Participant free and clear of any liens, claims, encumbrances or security interests, which, when valued at their Fair Market Value on the exercise date, have a value sufficient to pay the exercise price, provided that (1) at the time of exercise the Shares are publicly traded, (2) any remaining balance of the exercise price not satisfied by such delivery is paid by the Participant in cash or other permitted form of payment, (3) such delivery would not violate any Applicable Laws or agreement restricting the redemption of the Shares, (4) if required by the Administrator, any certificated Shares are endorsed or accompanied by an executed assignment separate from certificate, and (5) such Shares have been held by the Participant for any minimum period necessary to avoid adverse accounting treatment as a result of such delivery;
(iv) to the extent permitted by the Administrator at the time of exercise, except with respect to ISOs, surrendering the largest whole number of Shares then issuable upon the Option’s exercise which, when valued at their Fair Market Value on the exercise date, have a value sufficient to pay the exercise price, provided that (1) such Shares used to pay the exercise price will not be exercisable thereafter and (2) any remaining balance of the exercise price not satisfied by such net exercise is paid by the Participant in cash or other permitted form of payment;
(v) to the extent permitted by the Administrator at the time of exercise and permitted by Applicable Law, delivery of any other property that the Administrator determines is good and valuable consideration; or
(vi) to the extent permitted by the Company, any combination of the above payment forms approved by the Administrator.
(f) Non-Exempt U.S. Employees. No Option or Share Appreciation Right, whether or not vested, granted to an Employee who is a non-exempt employee for purposes of the U.S. Fair Labor Standards Act of 1938, as amended, will be first exercisable for any Shares until at least six months following the date of grant of such Award. Notwithstanding the foregoing, in accordance with the provisions of the U.S. Worker Economic Opportunity Act, any vested portion of such Award may be exercised earlier than six months following the date of grant of such Award in the event of (i) such Participant’s death or Disability, (ii) a Corporate Event in which such Award is not assumed, continued or substituted, (iii) a Change in Control, or (iv) such Participant’s retirement (as such term may be defined in the Award Agreement or another applicable agreement or, in the absence of any such definition, in accordance with the Company’s then current employment policies and guidelines). This Section 5(f) is intended to operate so that any income derived by a non-exempt employee in connection with the exercise or vesting of an Option or Share Appreciation Right will be exempt from his or her regular rate of pay.
6. RESTRICTED SHARES; RESTRICTED SHARE UNITS
(a) General. The Administrator may grant Restricted Shares, or the right to purchase Restricted Shares, to any Service Provider, subject to the Company’s right to repurchase all or part of such shares at their issue price or other stated or formula price from the Participant (or to require forfeiture of such shares) if conditions the Administrator specifies in the Award Agreement are not satisfied before the end of the applicable restriction period or periods that the Administrator establishes for such Award. In addition, the Administrator may grant to Service Providers Restricted Share Units, which may be subject to vesting, issuance and forfeiture conditions during the applicable restriction period or periods, as set forth in an Award Agreement. The Administrator will determine and set forth in the Award Agreement the terms and conditions for each Restricted Share and Restricted Share Unit Award, subject to the conditions and limitations contained in the Plan.
(b) Duration. Each Restricted Share or Restricted Share Unit will vest at such times and as specified in the Award Agreement, provided that the vesting schedule of a Restricted Share or Restricted Share Unit will not exceed ten years. Notwithstanding the foregoing, if the Participant, prior to the vesting date of a Restricted Share or Restricted Share Unit, violates the non-competition, non-solicitation, confidentiality or other similar restrictive covenant provisions of any employment contract, confidentiality and nondisclosure agreement or other agreement between the Participant and the Company or any of its Subsidiaries, the right of the Participant and the Participant’s transferees to receive Shares on the vesting of the Restricted Share or Restricted Share Unit issued to the Participant shall terminate immediately upon such violation, unless the Company otherwise determines. In addition, if, prior to the vesting date of a Restricted Share or Restricted Share Unit, the Participant is given notice by the Company or any of its Subsidiaries of the Participant’s Termination of Service by the Company or any of its Subsidiaries for Cause, and the effective date of such Termination of Service is subsequent to the date of the delivery of such notice, the right of the Participant and the Participant’s transferees to receive Shares as a result of the vesting of the Restricted Share or Restricted Share Unit issued to the Participant shall be suspended from the time of the delivery of such notice until the earlier of (i) such time as it is determined or otherwise agreed that the Participant’s service as a Service Provider will not be terminated for Cause as provided in such notice or (ii) the effective date of the Participant’s Termination of Service by the Company or any of its Subsidiaries for Cause (in which case the right of the Participant and the Participant’s transferees to receive Shares on the vesting of the Restricted Share or Restricted Share Unit issued to the Participant will terminate immediately upon the effective date of such Termination of Service).
(c) Dividends and Dividend Equivalents. Dividends or dividend equivalents may be paid or credited, as applicable, with respect to any Restricted Shares or Shares subject to Restricted Share Units, as determined (and on such terms as may be determined) by the Administrator and specified in the Award Agreement.
(d) Restricted Shares.
(i) Form of Award. The Company may require that the Participant deposit in escrow with the Company (or its designee) any certificates issued in respect of Restricted Shares, together with a stock transfer form endorsed in blank. Unless otherwise determined by the Administrator, a Participant will have voting and other rights as a shareholder of the Company with respect to any Restricted Shares.
(ii) Consideration. Restricted Shares may be granted in consideration for (A) cash or check, bank draft or money order payable to the Company, (B) past services to the Company or a Subsidiary, or (C) any other form of consideration (including future services) as the Administrator may determine to be acceptable and which is permissible under Applicable Laws.
(e) Restricted Share Units.
(i) Settlement. The Administrator may provide that settlement of Restricted Share Units will occur upon or as soon as reasonably practicable after the Restricted Share Units vest or will instead be deferred, on a mandatory basis or at the Participant’s election.
(ii) Shareholder Rights. A Participant will have no rights of a shareholder with respect to Shares subject to any Restricted Share Unit unless and until the Shares are delivered in settlement of the Restricted Share Unit.
(iii) Consideration. Unless otherwise determined by the Administrator at the time of grant, Restricted Share Units will be granted in consideration for the Participant’s services to the Company or a Subsidiary, such that the Participant will not be required to make any payment to the Company (other than such services) with respect to the grant or vesting of the Award, or the issuance of any Shares pursuant to the Award. If, at the time of grant, the Administrator determines that any consideration must be paid by the Participant (in a form other than the Participant’s services to the Company or a Subsidiary) upon the issuance of any Shares in settlement of the Award, such consideration may be paid in any form of consideration as the Administrator may determine to be acceptable and which is permissible under Applicable Laws.
7. OTHER SHARE BASED AWARDS
Other Share Based Awards may be granted to Participants, including Awards entitling Participants to receive Shares to be delivered in the future (whether based on specified performance criteria, performance goals or otherwise), in each case subject to any conditions and limitations in the Plan. Such Other Share Based Awards will also be available as a payment form in the settlement of other Awards, as standalone payments and as payment in lieu of compensation to which a Participant is otherwise entitled. Other Share Based Awards may be paid in Shares or other property, as the Administrator determines. Subject to the provisions of the Plan, the Administrator will determine the terms and conditions of each Other Share Based Award, including any purchase price, performance condition, performance goal, transfer restrictions, and vesting conditions, which will be set forth in the applicable Award Agreement.
8. ADJUSTMENTS FOR CHANGES IN SHARES AND CERTAIN OTHER EVENTS
(a) Equity Restructuring. In connection with any Equity Restructuring, notwithstanding anything to the contrary in this Section 8, the Administrator will equitably adjust (i) class(es) and maximum number of Shares subject to the Plan, (ii) the class(es) and maximum number of Shares that may be issued pursuant to the exercise of ISOs under Section 4(d) above and (iii) each outstanding Award as it deems appropriate to reflect the Equity Restructuring, which may include adjusting the number and type of securities subject to each outstanding Award and/or the Award’s exercise price or grant price (if applicable), granting new Awards to Participants, and making a cash payment to Participants. The adjustments provided under this Section 8(a) will be nondiscretionary and final and binding on the affected Participant and the Company; provided that the Administrator will determine whether an adjustment is equitable. Any adjustment made pursuant to this Section 8(a) to an Award held by a Participant subject to tax in the United States shall be made in a manner that complies with Section 409A and other Applicable Law.
(b) Corporate Events. In the event of any reorganization, merger, consolidation, combination, amalgamation, repurchase, recapitalization, liquidation, dissolution, or sale, transfer, exchange or other disposition of all or substantially all of the assets of the Company, or sale or exchange of Shares or other securities of the Company or a Change in Control (any “Corporate Event”), the Administrator, on such terms and conditions as it deems appropriate, is hereby authorized to take any one or more of the following actions whenever the Administrator determines that such action is appropriate:
(i) To provide for the cancellation of any such Award in exchange for either an amount of cash or other property with a value equal to the amount that could have been obtained upon the exercise or settlement of the vested portion of such Award or realization of the Participant’s rights under the vested portion of such Award, as applicable; provided that, if the amount that could have been obtained upon the exercise or settlement of the vested portion of such Award or realization of the Participant’s rights, in any case, is equal to or less than zero (as determined by the Administrator in its discretion), then the Award may be terminated without payment. In addition, such payments under this provision may, in the Administrator’s discretion, be delayed to the same extent that payment of consideration to the holders of Shares in connection with the Corporate Event is delayed as a result of escrows, earn outs, holdbacks or any other contingencies;
(ii) To provide that such Award shall vest and, to the extent applicable, be exercisable as to all Shares covered thereby, notwithstanding anything to the contrary in the Plan or the provisions of such Award as of a date prior to the effective time of such Corporate Event as the Administrator determines (or, if the Administrator does not determine such a date, as of the date that is five (5) days prior to the effective date of the Corporate Event), with such Award terminating if not exercised (if applicable) at or prior to the effective time of the Corporate Event; provided, however, that the Administrator may require Participants to complete and deliver to the Company a notice of exercise before the effective date of a Corporate Event, which exercise is contingent upon the effectiveness of such Corporate Event.
(iii) To provide that such Award be assumed by the successor or survivor corporation, or a parent or Subsidiary thereof, or shall be substituted for by awards covering the equity securities of the successor or survivor corporation, or a parent or Subsidiary thereof, with appropriate adjustments as to the number and kind of shares and/or applicable exercise or purchase price, in all cases, as determined by the Administrator;
(iv) To arrange for the assignment of any reacquisition or repurchase rights held by the Company in respect of Shares issued pursuant to the Award to the surviving corporation or acquiring corporation (or the surviving or acquiring corporation’s parent company);
(v) To arrange for the lapse, in whole or in part, of any reacquisition or repurchase rights held by the Company with respect to the Award;
(vi) To replace such Award with other rights or property selected by the Administrator; and/or
(vii) To provide that the Award will terminate and cannot vest, be exercised or become payable after the applicable transaction or event.
The Administrator need not take the same action or actions with respect to all Awards or portions thereof or with respect to all Participants. The Administrator may take different actions with respect to the vested and unvested portions of an Award.
(c) Administrative Stand Still. In the event of any pending Corporate Event or other similar transaction, for administrative convenience, the Administrator may refuse to permit the exercise of any Award for up to thirty days before or after such Corporate Event or other similar transaction.
(d) General. Except as expressly provided in the Plan or the Administrator’s action under the Plan, no Participant will have any rights due to any subdivision or consolidation of Shares of any class, dividend payment, increase or decrease in the number of Shares of any class, issue, rights issue, offer or dissolution, liquidation, merger, or consolidation of the Company or other corporation. Except as expressly provided with respect to an Equity Restructuring under Section 8(a) above or the Administrator’s action under the Plan, no issuance by the Company of Shares of any class, or securities convertible into Shares of any class, will affect, and no adjustment will be made regarding, the number of Shares subject to an Award or the Award’s grant or exercise price. The existence of the Plan, any Award Agreements and the Awards granted hereunder will not affect or restrict in any way the Company’s right or power to make or authorize (i) any adjustment, recapitalization, reorganization or other change in the Company’s capital structure or its business, (ii) any Corporate Event or (iii) sale or issuance of securities, including securities with rights superior to those of the Shares or securities convertible into or exchangeable for Shares. The Administrator may treat Participants and Awards (or portions thereof) differently under this Section 8.
9. GENERAL PROVISIONS APPLICABLE TO AWARDS
(a) Transferability. Except as the Administrator may determine or provide in an Award Agreement or otherwise for Awards, Awards may not be sold, assigned, transferred, pledged or otherwise encumbered, either voluntarily or by operation of law, except by will or the laws of descent and distribution, and, during the life of the Participant, will be exercisable only by the Participant. Notwithstanding the foregoing, the Administrator may, in its sole discretion, permit transfer of an Award pursuant to a domestic relations order or in such other manner that is not prohibited by applicable tax and securities laws upon the Participant’s request and provided that the Participant and the transferee enter into a transfer and other agreements as required by the Company. If an Option is an ISO, such Option may be deemed to be a Non-Qualified Option as a result of a transfer pursuant to this Section. References to a Participant, to the extent relevant in this context, will include references to a Participant’s authorized transferee that the Administrator specifically approves.
(b) Documentation. Each Award will be evidenced in an Award Agreement, which may be written or electronic, as the Administrator determines. By accepting any Award the Participant consents to receive documents by electronic delivery and to participate in the Plan through any on-line electronic system established and maintained by the Company or another third party selected by the Company. Each Award may contain terms and conditions in addition to (or a variation of or effecting a disapplication of) those set forth in the Plan. Any reference herein or in an Award Agreement to a “written” agreement or document will include any agreement or document delivered electronically, filed publicly at www.sec.gov (or any successor website thereto) or posted on the Company’s intranet (or other shared electronic medium controlled by the Company to which the Participant has access). As a condition to accepting an Award under the Plan, the Participant agrees to execute any additional documents or instruments necessary or desirable, as determined in the Administrator’s sole discretion, to carry out the purposes or intent of the Award, or facilitate compliance with securities and/or other regulatory requirements, in each case at the Administrator’s request.
(c) Discretion. Except as the Plan otherwise provides, each Award may be made alone or in addition or in relation to any other Award. The terms of each Award to a Participant need not be identical, and the Administrator need not treat Participants or Awards (or portions thereof) uniformly.
(d) Termination of Status. The Administrator will determine how the disability, death, retirement, authorized leave of absence or any other change or purported change in a Participant’s Service Provider status (including a change which would result in a Termination of Service under the Plan but not under the Non-Employee Sub-Plan or vice versa) affects an Award and the extent to which, and the period during which, the Participant, the Participant’s legal representative, conservator, guardian or Designated Beneficiary may exercise rights under the Award, if applicable.
(e) Withholding. Each Participant must pay the Company, or make provision satisfactory to the Administrator for payment of, any taxes (which includes any social security contributions or the like including but not limited to, if applicable, all liability to primary (employee) national insurance contributions) required by law to be withheld or paid by the Company or by any Subsidiary that is the employing entity of the Participant or which Participant has agreed to pay in connection with such Participant’s Awards by the date of the event creating the tax liability. A Participant may not be able to exercise an Award even though the Award is vested, and the Company shall have no obligation to issue Shares subject to an Award, unless and until such obligations are satisfied. The Company may deduct an amount sufficient to satisfy such tax obligations based on the maximum statutory withholding rates (or such other rate as may be determined by the Company after considering any accounting consequences or costs and Applicable Law) from any payment of any kind otherwise due to a Participant. To the extent permitted by the terms of an Award Agreement and subject to any Company insider trading, window period and/or dealing policy (including blackout periods), Participants may satisfy such tax obligations (i) in cash, by wire transfer of immediately available funds, by check made payable to the order of the Company, provided that the Company may limit the use of the foregoing payment forms if one or more of the payment forms below is permitted, (ii) to the extent permitted by the Administrator, in whole or in part by delivery of Shares, including Shares retained from the Award creating the tax obligation, valued at their Fair Market Value, (iii) if there is a public market for Shares at the time the tax obligations are satisfied, unless the Company otherwise determines, (A) delivery (including telephonically to the extent permitted by the Company) of an irrevocable and unconditional undertaking by a broker acceptable to the Company to deliver promptly to the Company sufficient funds to satisfy the tax obligations, or (B) delivery by the Participant to the Company of a copy of irrevocable and unconditional instructions to a broker acceptable to the Company to deliver promptly to the Company cash or a check sufficient to satisfy the tax and/or social security withholding, provided that such amount is paid to the Company at such time as may be required by the Administrator, (iv) withholding cash from an Award settled in cash, (v) withholding payment from any amounts otherwise payable to the Participant or (vi) to the extent permitted by the Company, any combination of the foregoing payment forms approved by the Administrator.
(f) Withholding Indemnification. As a condition to accepting an Award under the Plan, in the event that the amount of the Company’s and/or any Subsidiary’s withholding obligation in connection with such Award was greater than the amount actually withheld by the Company and/or its Subsidiaries, each Participant agrees to indemnify and hold the Company and/or its Subsidiaries harmless from any failure by the Company and/or its Subsidiaries to withhold the proper amount.
(g) Amendment of Award; Repricing. The Administrator may amend, modify or terminate any outstanding Award, including by cancelling and substituting another Award of the same or a different type, reducing the exercise price, changing the exercise or settlement date, converting an ISO to a Non-Qualified Option, taking any other action that is treated as a repricing under generally accepted accounting principles or by amending, waiving or relaxing any applicable performance criteria or goal(s). The Participant’s consent to such action will be required unless (i) the action, taking into account any related action, does not Materially Impair the Participant’s rights under the Award, or (ii) the change is permitted under Section 8 or pursuant to Section 10(f).
(h) Conditions on Delivery of Shares. The Company will not be obligated to deliver any Shares under the Plan or remove restrictions from Shares previously delivered under the Plan until (i) all Award conditions have been met or removed to the Company’s satisfaction, (ii) as determined by the Company, all other legal matters regarding the issuance and delivery of such Shares (including payment of nominal value) have been satisfied, including any applicable securities laws and stock exchange or stock market rules and regulations, and (iii) the Participant has executed and delivered to the Company such representations or agreements as the Administrator deems necessary or appropriate to satisfy any Applicable Laws. The Company’s inability to obtain authority from any regulatory body having jurisdiction, which the Administrator determines is necessary to the lawful issuance and sale of any securities, will relieve the Company of any liability for failing to issue or sell such Shares as to which such requisite authority has not been obtained.
(i) Acceleration. The Administrator may at any time provide that any Award will become immediately vested and fully or partially exercisable, free of some or all restrictions or conditions, or otherwise fully or partially realizable.
10. MISCELLANEOUS
(a) No Right to Employment or Other Status. No person will have any claim or right to be granted an Award, and the grant of an Award will not be construed as giving a Participant the right to continued employment or any other relationship with the Company. The Company expressly reserves the right at any time to dismiss or otherwise terminate its relationship with a Participant free from any liability or claim under the Plan or any Award, except as expressly provided in an Award Agreement. Further, nothing in the Plan, any Award Agreement or any other instrument executed thereunder or in connection with any Award will constitute any promise or commitment by the Company or a Subsidiary regarding the fact or nature of future positions, future work assignments, future compensation or any other term or condition of employment or service or confer any right or benefit under the Award or the Plan unless such right or benefit has specifically accrued under the terms of the Award Agreement and/or Plan.
(b) No Rights as Shareholder; Certificates. Subject to the Award Agreement, no Participant or Designated Beneficiary will have any rights as a shareholder with respect to any Shares to be distributed under an Award until becoming the record holder of such Shares. Notwithstanding any other provision of the Plan, unless the Administrator otherwise determines or Applicable Laws require, the Company will not be required to deliver to any Participant certificates evidencing Shares issued in connection with any Award and instead such Shares may be recorded in the books of the Company (or, as applicable, its transfer agent or stock plan administrator). The Company may place legends on certificates issued under the Plan that the Administrator deems necessary or appropriate to comply with Applicable Laws.
(c) Effective Date and Term of Plan. The Plan will come into existence on the day it is adopted by the Board but no Awards may be granted under the Plan prior to the Effective Date. Unless earlier terminated by the Board, the Plan will remain in effect until the tenth anniversary of the Effective Date, but Awards previously granted may extend beyond that date in accordance with the Plan. No ISOs may be granted after the tenth anniversary of the earlier of (i) the date the Plan is adopted by the Board, or (ii) the date the Plan is approved by the Company’s shareholders. If the Plan is not approved by the Company’s shareholders within 12 months of the date of Board approval of the Plan, all ISOs will be treated as Non-Qualified Options.
(d) Amendment and Termination of Plan. The Administrator may amend, suspend or terminate the Plan at any time; provided that no amendment, suspension or termination may Materially Impair any Award outstanding at the time of such amendment without the affected Participant’s written consent. No Awards may be granted under the Plan during any suspension period or after Plan termination. Awards outstanding at the time of any Plan suspension or termination will continue to be governed by the Plan and the Award Agreement, as in effect before such suspension or termination. The Board will obtain shareholder approval of any Plan amendment to the extent necessary to comply with Applicable Laws.
(e) Provisions for Foreign Participants. The Administrator may modify Awards granted to Participants who are nationals of, or employed in, a jurisdiction outside the United Kingdom and the United States or establish subplans or procedures under the Plan to address differences in laws, rules, regulations or customs of such international jurisdictions with respect to tax, securities, currency, employee benefit or other matters, including as may be necessary or appropriate in the Administrator’s discretion to grant Awards under any tax-favourable regime that may be available in any jurisdiction (provided that Administrator approval will not be necessary for immaterial modifications to the Plan or any Award Agreement to ensure or facilitate compliance with the laws of the relevant foreign jurisdiction).
(f) Section 409A. The following provisions only apply to Participants subject to tax in the United States:
(i) General. The Company intends that all Awards be structured to comply with, or be exempt from, Section 409A, such that no adverse tax consequences, interest, or penalties under Section 409A apply. Notwithstanding anything in the Plan or any Award Agreement to the contrary, the Administrator may, without a Participant’s consent, amend this Plan or Awards, adopt policies and procedures, or take any other actions (including amendments, policies, procedures and retroactive actions) as are necessary or appropriate to preserve the intended tax treatment of Awards, including any such actions intended to (A) exempt this Plan or any Award from Section 409A, or (B) comply with Section 409A, including regulations, guidance, compliance programs and other interpretative authority that may be issued after an Award’s grant date. The Company makes no representations or warranties as to an Award’s tax treatment under Section 409A or otherwise. The Company will have no obligation under this Section 10(f) or otherwise to avoid the taxes, penalties or interest under Section 409A with respect to any Award and will have no liability to any Participant or any other person if any Award, compensation or other benefits under the Plan are determined to constitute noncompliant “nonqualified deferred compensation” subject to taxes, penalties or interest under Section 409A.
(ii) Separation from Service. If an Award constitutes “nonqualified deferred compensation” under Section 409A, any payment or settlement of such Award upon a termination of a Participant’s Service Provider relationship will, to the extent necessary to avoid taxes under Section 409A, be made only upon the Participant’s “separation from service” (within the meaning of Section 409A), whether such “separation from service” occurs upon or after the termination of the Participant’s Service Provider relationship. For purposes of this Plan or any Award Agreement relating to any such payments or benefits, references to a “termination,” “termination of service”, “termination of employment” or like terms means a “separation from service.”
(iii) Payments to Specified Employees. Notwithstanding any contrary provision in the Plan or any Award Agreement, any payment(s) of “nonqualified deferred compensation” required to be made under an Award to a “specified employee” (as defined under Section 409A and as the Administrator determines) due to his or her “separation from service” will, to the extent necessary to avoid taxes under Section 409A(a)(2)(B)(i) of the Code, be delayed for the six-month period immediately following such “separation from service” (or, if earlier, until the specified employee’s death) and will instead be paid (as set forth in the Award Agreement) on the day immediately following such six-month period or as soon as administratively practicable thereafter (without interest). Any payments of “nonqualified deferred compensation” under such Award payable more than six months following the Participant’s “separation from service” will be paid at the time or times the payments are otherwise scheduled to be made.
(g) 10% Shareholders. The Administrator may grant ISOs only to employees of the Company, any of its present or future parent or subsidiary corporations, as defined in Sections 424(e) or (f) of the Code, respectively, and any other entities the employees of which are eligible to receive ISOs under the Code. If an ISO is granted to a Greater Than 10% Shareholder, the exercise price will not be less than 110% of the Fair Market Value on the Option’s grant date, and the term of the Option will not exceed five years. All ISOs will be subject to and construed consistently with Section 422 of the Code. By accepting an ISO, the Participant agrees to give prompt notice to the Company of dispositions or other transfers (other than in connection with a Change in Control) of Shares acquired under the Option made within (i) two years from the grant date of the Option or (ii) one year after the transfer of such Shares to the Participant, specifying the date of the disposition or other transfer and the amount the Participant realized, in cash, other property, assumption of indebtedness or other consideration, in such disposition or other transfer. Neither the Company nor the Administrator will be liable to a Participant, or any other party, if an ISO fails or ceases to qualify as an “incentive stock option” under Section 422 of the Code. Any ISO or portion thereof that fails to qualify as an “incentive stock option” under Section 422 of the Code for any reason, including becoming exercisable with respect to Shares having a fair market value exceeding the $100,000 limitation under Treasury Regulation Section 1.422-4, will be a Non-Qualified Option.
(h) Limitations on Liability. Notwithstanding any other provisions of the Plan, no individual acting as a director, officer, other employee or agent of the Company or any Subsidiary will be liable to any Participant, former Participant, spouse, beneficiary, or any other person for any claim, loss, liability, or expense incurred in connection with the Plan or any Award, and such individual will not be personally liable with respect to the Plan because of any contract or other instrument executed in his or her capacity as an Administrator, director, officer, other employee or agent of the Company or any Subsidiary. As a condition to accepting an Award under the Plan, each Participant (i) agrees to not make any claim against the Company, the Group or any of its officers, Directors, Employees or Subsidiaries related to tax or social security liabilities arising from such Award or other Company or Group compensation and (ii) acknowledges that such Participant was advised to consult with his or her own personal tax, financial and other legal advisors regarding the tax and social security consequences of the Award and has either done so or knowingly and voluntarily declined to do so. The Company will indemnify and hold harmless each director, officer, other employee and agent of the Company or any Subsidiary that has been or will be granted or delegated any duty or power relating to the Plan’s administration or interpretation, against any cost or expense (including attorneys’ fees) or liability (including any sum paid in settlement of a claim with the Administrator’s approval) arising from any act or omission concerning this Plan unless arising from such person’s own fraud or bad faith.
(i) No Obligation to Notify or Minimize Taxes. Except as required by Applicable Laws the Company has no duty or obligation to any Participant to advise such Participant as to the time or manner of exercising such Award. Furthermore, the Company has no duty or obligation to warn or otherwise advise such Participant of a pending termination or expiration of an Award or a possible period in which the Award may not be exercised. The Company has no duty or obligation to minimize the tax or social security consequences of an Award to the holder of such Award and will not be liable to any holder of an Award for any adverse tax or social security consequences to such holder in connection with an Award.
(j) Data Privacy.
(i) As a condition for receiving any Award, each Participant acknowledges that the Company and any Subsidiary may collect, use and transfer, in electronic or other form, personal data as described in this section by and among the Company and its Subsidiaries and affiliates exclusively for implementing, administering and managing the Participant’s participation in the Plan. The Company (as above) may hold certain personal information about a Participant, including the Participant’s name, address and telephone number; birthdate; social security, insurance number or other identification number; salary; nationality; job title(s); any Shares held in the Company (as above); and Award details, to implement, manage and administer the Plan and Awards (the “Data”). The Company (as above) may transfer the Data amongst themselves as necessary to implement, administer and manage a Participant’s participation in the Plan, and the Company (as above) may transfer the Data to third parties assisting the Company with Plan implementation, administration and management. These recipients may be located in the Participant’s country, or elsewhere, and the Participant’s country may have different data privacy laws and protections than the recipients’ country. By accepting an Award, each Participant acknowledges that such recipients may receive, possess, use, retain and transfer the Data, in electronic or other form, to implement, administer and manage the Participant’s participation in the Plan, including any required Data transfer to a broker or other third party with whom the Company or the Participant may elect to deposit any Shares. The Data related to a Participant will be held only as long as necessary to implement, administer, and manage the Participant’s participation in the Plan. A Participant may, at any time, view the Data that the Company holds regarding such Participant, request additional information about the storage and processing of the Data regarding such Participant and recommend any necessary corrections to the Data regarding the Participant in writing, without cost, by contacting the local human resources representative.
(ii) For the purpose of operating the Plan in the European Union, Switzerland and the United Kingdom, the Company will collect and process information relating to Participants in accordance with the privacy notice which is provided to each Participant.
(k) Severability. If any portion of the Plan or any Award Agreement or any action taken thereunder is held illegal or invalid for any reason, the illegality or invalidity will not affect the remaining parts of the Plan or such Award Agreement, and the Plan and such Award Agreement will be construed and enforced as if the illegal or invalid provisions had been excluded, and the illegal or invalid action will be null and void.
(l) Governing Documents. If any contradiction occurs between the Plan and any Award Agreement or other written agreement between a Participant and the Company (or any Subsidiary) that the Administrator has approved, the Plan will govern, unless it is expressly specified in such Award Agreement or other written document that a specific provision of the Plan will not apply.
All Awards will be subject to Applicable Laws on insider trading and dealing and any specific insider trading, window period and/or dealing policy adopted by the Company.
(m) Governing Law and Jurisdiction. The Plan and all Awards, including any non-contractual obligations arising in connection therewith, will be governed by and interpreted in accordance with the laws of England and Wales, disregarding any jurisdiction’s choice-of-law principles requiring the application of a jurisdiction’s laws other than that of England and Wales and the courts of England and Wales shall have exclusive jurisdiction to hear any dispute.
(n) Claw-back Provisions. All Awards (including any proceeds, gains or other economic benefit the Participant actually or constructively receives upon receipt or exercise of any Award or the receipt or resale of any Shares underlying the Award) will be subject to any Company claw-back policy that may be adopted from time to time to the extent such policy applies to the relevant Participant, including any claw-back policy adopted to comply with Applicable Laws (including the Dodd-Frank Wall Street Reform and Consumer Protection Act and any rules or regulations promulgated thereunder) as set forth in such claw-back policy or the Award Agreement , to the extent applicable and permissible under Applicable Laws. No recovery of compensation under such a claw-back policy will be an event giving rise to a Participant’s right to voluntary terminate employment upon a “resignation for good reason,” or for a “constructive termination” or any similar term under any plan of or agreement with the Company.
(o) Other Group Company policies. All Awards (including any proceeds, gains or other economic benefit the Participant actually or constructively receives upon receipt or exercise of any Award or the receipt or resale of any Shares underlying the Award) will be subject to any relevant Company or Group Company policy to the extent such policy applies to the relevant Participant, including but not limited to any remuneration policy and/or share retention, ownership, or holding policy that may be adopted from time to time.
(p) Titles and Headings. The titles and headings in the Plan are for convenience of reference only and, if any conflict, the Plan’s text, rather than such titles or headings, will control.
(q) Conformity to Applicable Laws. Participant acknowledges that the Plan is intended to conform to the extent necessary with Applicable Laws. Notwithstanding anything herein to the contrary, the Plan and all Awards will be administered only in conformance with Applicable Laws. To the extent Applicable Laws permit, the Plan and all Award Agreements will be deemed amended as necessary to conform to Applicable Laws and may be unilaterally cancelled by the Company (with the effect that all Participant’s rights thereunder lapse with immediate effect) if the Administrator determines in its reasonable discretion that such conformity is not possible or practicable.
(r) Relationship to Other Benefits. No payment under the Plan will be taken into account in determining any benefits under any pension, retirement, savings, profit sharing, group insurance, welfare or other benefit plan of the Company or any Subsidiary except as expressly provided in writing in such other plan or an agreement thereunder.
(s) Broker-Assisted Sales. In the event of a broker-assisted sale of Shares in connection with the payment of amounts owed by a Participant under or with respect to the Plan or Awards: (a) any Shares to be sold through the broker-assisted sale will be sold (subject in all cases to the Administrator having regard to the orderly marketing and disposal of such Shares, and having the discretion to delay broker-assisted sales for such reasons) on the day the payment first becomes due, or as soon thereafter as practicable; (b) such Shares may be sold as part of a block trade with other Participants in the Plan in which all Participants receive an average price; (c) the applicable Participant will be responsible for all broker’s fees and other costs of sale, and by accepting an Award, each Participant agrees to indemnify and hold the Company harmless from any losses, costs, damages, or expenses relating to any such sale; (d) to the extent the Company or its designee receives proceeds of such sale that exceed the amount owed, the Company will pay such excess in cash to the applicable Participant as soon as reasonably practicable; (e) the Company and its designees are under no obligation to arrange for such sale at any particular price; and (f) in the event the proceeds of such sale are insufficient to satisfy the Participant’s applicable obligation, the Participant may be required to pay immediately upon demand to the Company or its designee, or the Company or any Subsidiary may withhold from any payment to be made to the Participant (including but not limited to that Participant’s salary), an amount in cash sufficient to satisfy any remaining portion of the Participant’s obligation.
(t) Change in Time Commitment. In the event a Participant’s regular level of time commitment in the performance of his or her services for the Company and any Subsidiary is reduced (for example, and without limitation, if the Participant is an Employee of the Company and the Employee has a change in status from a full-time Employee to a part-time Employee or takes an extended leave of absence) after the date of grant of any Award to the Participant, the Administrator may determine, to the extent permitted by Applicable Laws, to (i) make a corresponding reduction in the number of shares or cash amount subject to any portion of such Award that is scheduled to vest or become payable after the date of such change in time commitment, and (ii) in lieu of or in combination with such a reduction, extend the vesting or payment schedule applicable to such Award. In the event of any such reduction, the Participant will have no right with respect to any portion of the Award that is so reduced or extended.
(u) Deferrals. To the extent permitted by Applicable Laws, the Administrator, in its sole discretion, may determine that the delivery of Shares or the payment of cash, upon the exercise, vesting or settlement of all or a portion of any Award may be deferred and may also establish programs and procedures for deferral elections to be made by Participants. Deferrals will be made in accordance with the requirements of Section 409A to the extent applicable.
11. Covenants of the Company.
(a) Compliance with Law. The Company will seek to obtain from each regulatory commission or agency, as may be deemed to be necessary, having jurisdiction over the Plan such authority as may be required to grant Awards and to issue and sell Shares upon exercise or vesting of the Awards; provided, however, that this undertaking will not require the Company to register under the Securities Act the Plan, any Award or any Shares issued or issuable pursuant to any such Award. If, after reasonable efforts and at a reasonable cost, the Company is unable to obtain from any such regulatory commission or agency the authority that counsel for the Company deems necessary or advisable for the lawful issuance and sale of Shares under the Plan, the Company will be relieved from any liability for failure to issue and sell Shares upon exercise or vesting of such Awards unless and until such authority is obtained. A Participant is not eligible for the grant of an Award or the subsequent issuance of Shares pursuant to the Award if such grant or issuance would be in violation of any Applicable Laws.
12. DEFINITIONS.
As used in the Plan, the following words and phrases will have the following meanings:
(a) “ADSs” means American Depositary Shares, representing Ordinary Shares on deposit with a U.S. banking institution selected by the Company and which are registered pursuant to a Form F-6.
(b) “Administrator” means the Board or a Committee to the extent that the Board’s powers or authority under the Plan have been delegated to such Committee.
(c) “Applicable Laws” means any applicable laws, statutes, constitutions, principles of common law, resolutions, ordinances, codes, edicts, decrees, rules, listing rules, regulations, judicial decisions, rulings or requirements issued, enacted, adopted, promulgated, implemented or otherwise put into effect by or under the authority of any Governmental Body (including under the authority of any applicable self-regulating organization such as the Nasdaq Stock Market, New York Stock Exchange, or the Financial Industry Regulatory Authority), including without limitation: (a) the requirements relating to the administration of equity incentive plans under English, U.S. federal and state securities, tax and other applicable laws, rules and regulations, the applicable rules of any stock exchange or quotation system on which the Shares are listed or quoted and the applicable laws and rules of any other country or jurisdiction where Awards are granted; and (b) corporate, securities, tax or other laws, statutes, rules, requirements or regulations, whether U.S. federal, state, local or foreign, applicable in the United Kingdom, United States or any other relevant jurisdiction.
(d) “Award” means, individually or collectively, a grant under the Plan of Options, Share Appreciation Rights, Restricted Shares, Restricted Share Units, or Other Share Based Awards.
(e) “Award Agreement” means a written agreement between the Company and a Participant evidencing an Award, which may be electronic. The Award Agreement generally consists of the grant notice and the agreement that contains such terms and conditions as the Administrator determines, consistent with and subject to the terms and conditions of the Plan.
(f) “Board” means the Board of Directors of the Company (or its designee).
(g) “Cause” means (i) if a Participant is a party to a written employment or consulting agreement with the Company or any of its Subsidiaries or an Award Agreement in which the term “cause” is defined (a “Relevant Agreement”), “Cause” as defined in the Relevant Agreement, and (ii) if no Relevant Agreement exists, (A) the Administrator’s determination that the Participant failed to substantially perform the Participant’s duties (other than a failure resulting from the Participant’s Disability); (B) the Administrator’s determination that the Participant failed to carry out, or comply with any lawful directive of the Board or the Participant’s immediate supervisor; (C) the occurrence of any act or omission by the Participant that could reasonably be expected to result in (or has resulted in) the Participant’s conviction, plea of no contest, plea of nolo contendere, or imposition of unadjudicated probation for any felony or indictable offence or crime involving fraud, dishonesty or moral turpitude (or equivalent in any jurisdiction); (D) the Participant’s unlawful use (including being under the influence) or possession of illegal drugs on the premises of the Company or any of its Subsidiaries or while performing the Participant’s duties and responsibilities for the Company or any of its Subsidiaries; (E) the Participant’s commission of (or attempted commission of) an act of fraud, embezzlement, misappropriation, misconduct, or breach of fiduciary duty against the Company or any of its Subsidiaries; (F) the Participant’s unauthorized use or disclosure of the confidential information or trade secrets of the Company or any Subsidiary; or (G) the Participant’s material violation of any contract or agreement between the Participant and the Company (or Subsidiary) or of any statutory duty owed to the Company (or Subsidiary) or such Participant’s material failure to comply with the written policies or rules of the Company (or Subsidiary).
(h) “Change in Control” means and includes each of the following:
(i) a Sale; or
(ii) a Takeover.
The Administrator shall have full and final authority, which shall be exercised in its sole discretion, to determine conclusively whether a Change in Control has occurred pursuant to the above definition, the date of the occurrence of such Change in Control and any incidental matters relating thereto; provided that any exercise of authority in conjunction with a determination of whether a Change in Control is a “change in control event” as defined in Treasury Regulation Section 1.409A-3(i)(5) shall be consistent with such regulation.
Notwithstanding the foregoing or any other provision of this Plan, the term Change in Control shall not include a sale of assets, merger or other transaction effected exclusively for the purpose of changing the domicile of the Company.
(i) “Code” means the US Internal Revenue Code of 1986, as amended, and the regulations issued thereunder.
(j) “Committee” means one or more committees or subcommittees of the Board, which may include one or more Company directors or executive officers, to the extent Applicable Laws permit. To the extent required to comply with the provisions of Rule 16b-3, it is intended that each member of the Committee will be, at the time the Committee takes any action with respect to an Award that is subject to Rule 16b-3, a “non-employee director” within the meaning of Rule 16b-3; however, a Committee member’s failure to qualify as a “non-employee director” within the meaning of Rule 16b-3 will not invalidate any Award granted by the Committee that is otherwise validly granted under the Plan.
(k) “Company” means Immunocore Holdings Plc, registered in England and Wales with company number 13119746, or any successor.
(l) “Control” has the meaning given in section 995(2) of the UK Income Tax Act 2007, unless otherwise specified.
(m) “Corporate Event” has the meaning given to it in Section 8(b).
(n) “Designated Beneficiary” means: (i) a Participant’s personal representative appointed on Participant’s death; or (ii) if the Administrator permits from time to time in its discretion, the beneficiary or beneficiaries a Participant designates, in a manner the Administrator determines, to receive amounts due or exercise the Participant’s rights if the Participant dies or becomes incapacitated.
(o) “Director” means a Board member.
(p) “Disability” means a permanent and total disability under Section 22(e)(3) of the Code, as amended, and will be determined by the Administrator on the basis of such medical evidence as the Administrator deems warranted under the circumstances.
(q) “Effective Date” means immediately prior to the IPO Date, provided this Plan is approved by the Company’s shareholders prior to the IPO Date.
(r) “Employee” means any employee of the Company or its Subsidiaries.
(s) “Equity Restructuring” means any return of capital (including a share dividend), bonus issue of shares or other Company securities by way of capitalization of profits, share split, reverse share split, spin-off, rights offering, re-designation, redenomination, consolidation recapitalization through a large, nonrecurring cash dividend, or any similar equity restructuring transaction, that affects the number or class of Shares (or other Company securities) or the nominal value of Shares (or other Company securities) and causes a change in the per share value of the Shares underlying outstanding Awards. Notwithstanding the foregoing, the conversion of any convertible securities of the Company will not be treated as an Equity Restructuring.
(t) “Exchange Act” means the US Securities Exchange Act of 1934, as amended, and the rules and regulations promulgated thereunder.
(u) “Fair Market Value” means, as of any date, unless otherwise determined by the Administrator, the value of the Shares (as determined on a per share or aggregate basis, as applicable) determined as follows:
(i) If the Shares are listed on any established stock exchange or traded on any established market, the Fair Market Value will be the closing sales price for such Shares as quoted on such exchange or market (or the exchange or market with the greatest volume of trading in the Shares) on the date of determination, as reported in a source the Administrator deems reliable.
(ii) If there is no closing sales price for the Shares on the date of determination, then the Fair Market Value will be the closing selling price on the last preceding date for which such quotation exists.
(iii) In the absence of such markets for the Shares, or if otherwise determined by the Administrator, the Fair Market Value will be determined by the Administrator in good faith and, for Participants subject to tax in the United States, in a manner that complies with Sections 409A and, if applicable, 422 of the Code.
(v) “Governmental Body” means any: (a) nation, state, commonwealth, province, territory, county, municipality, district or other jurisdiction of any nature; (b) United Kingdom, U.S. federal, state, local, municipal, foreign or other government; (c) governmental or regulatory body, or quasi-governmental body of any nature (including any governmental division, department, administrative agency or bureau, commission, authority, instrumentality, official, ministry, fund, foundation, center, organization, unit, body or entity and any court or other tribunal, and for the avoidance of doubt, any tax authority) or other body exercising similar powers or authority; or (d) self-regulatory organization (including the Nasdaq Stock Market, New York Stock Exchange, and the Financial Industry Regulatory Authority).
(w) “Greater Than 10% Shareholder” means an individual then owning (within the meaning of Section 424(d) of the Code) more than 10% of the total combined voting power of all classes of equity securities of the Company or its parent or subsidiary corporation, as defined in Section 424(e) and (f) of the Code, respectively.
(x) “Group” means the Company and its Subsidiaries (references to “Group Company” shall be construed accordingly).
(y) “IPO Date” means the date of the underwriting agreement between the Company and the underwriter(s) managing the initial public offering of the Company’s ADSs, pursuant to which the ADSs are priced for the initial public offering.
(z) “ISO” means an Option intended to be, and that qualifies as, an “incentive stock option” as defined in Section 422 of the Code.
(aa) “Materially Impair” means any amendment to the terms of the Award that materially adversely affects the Participant’s rights under the Award. A Participant's rights under an Award will not be deemed to have been Materially Impaired by any such amendment if the Administrator, in its sole discretion, determines that the amendment, taken as a whole, does not materially impair the Participant's rights. For example, the following types of amendments to the terms of an Award do not Materially Impair the Participant’s rights under the Award: (i) imposition of reasonable restrictions on the minimum number of shares subject to an Option that may be exercised; (ii) to maintain the qualified status of the Award as an ISO under Section 422 of the Code; (iii) to change the terms of an ISO in a manner that disqualifies, impairs or otherwise affects the qualified status of the Award as an ISO under Section 422 of the Code; (iv) to clarify the manner of exemption from, or to bring the Award into compliance with or qualify it for an exemption from, Section 409A; or (v) to comply with other Applicable Laws.
(bb) “Non-Employee Sub-Plan” means the Non-Employee Sub-Plan to the Plan adopted by the Board.
(cc) “Non-Qualified Option” means an Option not intended or not qualifying as an ISO.
(dd) “Option” means an option to purchase Shares.
(ee) “Ordinary Share” means an ordinary share of GBP 0.002 each in the capital of the Company.
(ff) “Other Share Based Awards” means awards of Shares, and other awards valued wholly or partially by referring to, or are otherwise based on, Shares or other property, including the appreciation in value thereof (e.g., options or share rights with an exercise price or strike price less than 100% of the Fair Market Value at the time of grant), that may be granted either alone or in addition to Awards provided for under Section 5 and Section 6.
(gg) “Participant” means a Service Provider who has been granted an Award.
(hh) “Plan” means this 2021 Equity Incentive Plan, as amended from time to time.
(ii) “Prior Plans” means (i) the Share Option Scheme (Incorporating Management Incentive Options) originally adopted by the UK Company in 2008; (ii) the Non Tax-Advantaged Share Option Plan originally adopted by the UK Company on 15 May 2015; (iii) the Company Share Option Plan originally adopted by the UK Company on 15 May 2015; (iv) the 2018 Non Tax-Advantaged Share Option Plan originally adopted by the UK Company on 14 August 2018; (v) the Non Tax-Advantaged Share Option Plan originally adopted by the UK Company on 20 April 2020; (vi) the Company Share Option Plan originally adopted by the UK Company on 20 April 2020; and (vii) the standalone Unapproved Share Option Agreements documenting options originally granted by the UK Company to individuals outside a plan prior to the Effective Date (each as subsequently amended from time to time and as assumed or adopted by the Company prior to the Effective Date).
(jj) “Quarter Date” means each of 1 January, 1 April, 1 July and 1 October.
(kk) “Restricted Shares” means Shares awarded to a Participant under Section 6 subject to certain vesting conditions and other restrictions.
(ll) “Restricted Share Unit” means an unfunded, unsecured right to receive, on the applicable settlement date, one Share (or, if specified in the Award Agreement, other consideration determined by the Administrator to be of equal value as of such settlement date), subject to certain vesting conditions and other restrictions provided that nothing contained in the Plan or any Award Agreement, and no action taken pursuant to its provisions, will create or be construed to create a trust of any kind or a fiduciary relationship between a Participant and the Company or a Subsidiary or any other person.
(mm) “Rule 16b-3” means Rule 16b-3 promulgated under the Exchange Act or any successor to Rule 16b-3, as in effect from time to time.
(nn) “Sale” means the sale of all or substantially all of the assets of the Company.
(oo) “Section 409A” means Section 409A of the Code and all regulations, guidance, compliance programs and other interpretative authority thereunder.
(pp) “Securities Act” means the US Securities Act of 1933, as amended.
(qq) “Service Provider” means an Employee, Director or Consultant, provided that Consultants and Directors who are not Employees are only considered “Service Providers” eligible to be granted Awards under the Non-Employee Sub-Plan.
(rr) “Share” means an Ordinary Share or the number of ADSs equal to an Ordinary Share.
(ss) “Share Appreciation Right” means a Share Appreciation right granted under Section 5.
(tt) “Share Reserve” has the meaning given to it in Section 4(a).
(uu) “Subsidiary” has the meaning as set out in section 1159 of the UK Companies Act 2006.
(vv) “Substitute Awards” means Awards granted or Shares issued by the Company in assumption of, or in substitution or exchange for, awards previously granted, or the right or obligation to make future awards, in each case by a company acquired by the Company or any Subsidiary or with which the Company or any Subsidiary combines.
(ww) “Takeover” means if any person (or a group of persons acting in concert) (the “Acquiring Person”):
(i) obtains Control of the Company as the result of making a general offer to:
(1) acquire all of the issued ordinary share capital of the Company, which is made on a condition that, if it is satisfied, the Acquiring Person will have Control of the Company; or
(2) acquire all of the shares in the Company which are of the same class as the Shares; or
(ii) obtains Control of the Company as a result of a compromise or arrangement sanctioned by a court under Section 899 of the UK Companies Act 2006, or sanctioned under any other similar law of another jurisdiction; or
(iii) becomes bound or entitled under Sections 979 to 985 of the UK Companies Act 2006 (or similar law of another jurisdiction) to acquire shares of the same class as the Shares; or
(iv) obtains Control of the Company in any other way.
(xx) “Termination of Service” means the date the Participant ceases to be a Service Provider as defined in the Plan.
(yy) “UK Company” means Immunocore Limited registered in England and Wales with company number 06456207.
NON-EMPLOYEE SUB-PLAN
TO THE Immunocore HOLDINGS PLC 2021 EQUITY INCENTIVE PLAN
This sub-plan (the “Non-Employee Sub-Plan”) to the Immunocore Holdings Plc 2021 Equity Incentive Plan (the “Plan”) governs the grant of Awards to Consultants (defined below) and Directors who are not Employees. The Non-Employee Sub-Plan incorporates all the provisions of the Plan except as modified in accordance with the provisions of this Non-Employee Sub-Plan.
Awards granted pursuant to the Non-Employee Sub-Plan are not granted pursuant to an “employees’ share scheme” for the purposes of UK legislation.
For the purposes of the Non-Employee Sub-Plan, the provisions of the Plan shall operate subject to the following modifications:
1. Interpretation
In the Non-Employee Sub-Plan, unless the context otherwise requires, the following words and expressions have the following meanings:
“Consultant” means any person, including any adviser, engaged by the Company or any Group Company to render services to such entity if the consultant or adviser: (i) renders bona fide services to the Company or any Group Company; (ii) renders services not in connection with the offer or sale of securities in a capital-raising transaction and does not directly or indirectly promote or maintain a market for the Company’s securities; and (iii) is a natural person. Notwithstanding the foregoing, a person is treated as a Consultant only if a Form S-8 Registration Statement under the Securities Act is available to register either the offer or the sale of the Company’s securities to such person.
“Service Provider” means a Consultant or Director who is not an Employee.
“Termination of Service” means, subject to Section 3 below, the date the Participant ceases to be a Service Provider as defined in this Non-Employee Sub-Plan.
2. Eligibility
Service Providers are eligible to be granted Awards under the Non-Employee Sub-Plan.
3. Service Provider status and Termination of Service
If the Administrator so determines, a Participant who ceases to be a Service Provider for the purposes of this Non-Employee Sub-Plan and who becomes a Service Provider as defined in the Plan immediately thereafter (provided that there is no interruption or termination of the Participant’s service with the Company or a Subsidiary) may be considered to remain continuously a Service Provider for the purposes of the Non-Employee Sub-Plan.
APPENDIX 2
OPTION GRANT NOTICE (US / UK)
IMMUNOCORE HOLDINGS PLC
2021 EQUITY INCENTIVE PLAN [:NON-EMPLOYEE SUB-PLAN]1
Capitalized terms not specifically defined in this Option Grant Notice (the “Grant Notice”) have the meanings given to them in the 2021 Equity Incentive Plan [:Non-Employee Sub-Plan]2 (as amended from time to time, the “Plan”) of Immunocore Holdings Plc (the “Company”).
The Company has granted to the participant listed below (“Participant”) the option described in this Grant Notice (the “Option”), subject to the terms and conditions of the Plan and the Option Agreement attached as Exhibit A (the “Agreement”), both of which are incorporated into this Grant Notice by reference.
| Participant: | |
| Grant Date: | |
| Exercise Price per Share: | |
| Shares Subject to the Option: | |
| Final Expiration Date: | The day before the [10th] anniversary of the Grant Date |
| Vesting Commencement Date: | |
| Vesting Schedule3: | [1/4 of the total number of Shares under Option shall vest and become exercisable on the first anniversary of the Vesting Commencement Date, and 1/12th of the remaining number of Shares under Option shall vest and become exercisable on each Quarter Date thereafter, subject to Participant remaining continuously a Service Provider as of each such date]. |
| Type of Option4 | [ISO]5[Non-Qualified Option6] |
By Participant’s signature below, Participant agrees to be bound by the terms of this Grant Notice, the Plan, the Agreement and any Group Company policy that may be applicable to the Participant and the Option from time to time (the “Policies”) [including but not limited to the [Company’s claw-back policy / share retention policy / remuneration policy]]7. Participant has reviewed the Plan, this Grant Notice, the Agreement and the Policies in their entirety, has had an opportunity to obtain the advice of counsel prior to executing this Grant Notice and fully understands all provisions of the Plan, this Grant Notice, the Agreement and the Policies. Participant hereby agrees to accept as binding, conclusive and final all decisions or interpretations of the Administrator upon any questions arising under the Plan, this Grant Notice or the Agreement.
1 Note to draft: For Consultants and Directors who are not Employees
2 Note to draft: For Consultants and Directors who are not Employees
3 Note to draft: Selection of applicable vesting schedule, or determination that a different vesting schedule shall apply, subject to discretion of Administrator.
4 If this is an ISO, it (plus other outstanding ISOs) cannot be first exercisable for more than $100,000 in value (measured by exercise price) in any calendar year. Any excess over $100,000 is a Non-Qualified Option.
5 Note to draft: Available only for US taxpayer employees.
6 Note to draft: For all other Service Providers.
7 Note to draft: Delete as applicable
By accepting this Option, Participant consents to receive this Grant Notice, the Agreement, the Plan, the Policies and any other Plan-related documents by electronic delivery and to participate in the Plan through an on-line or electronic system established and maintained by the Company or another third party designated by the Company. Counterparts may be delivered via facsimile, electronic mail (including pdf or any electronic signature complying with the US federal ESIGN Act of 2000, Uniform Electronic Transactions Act or other Applicable Law) or other transmission method and any counterpart so delivered will be deemed to have been duly and validly delivered and be valid and effective for all purposes.
| Immunocore HOLDINGS Plc | PARTICIPANT | ||
| By: | |||
| Name | [Participant Name] | ||
| Title: | |||
Exhibit A
OPTION AGREEMENT
Capitalized terms not specifically defined in this Agreement have the meanings specified in the Grant Notice or, if not defined in the Grant Notice, in the Plan.
| 1. | GENERAL |
| 1.1. | Grant of Option |
The Company has granted to Participant the Option effective as of the grant date set forth in the Grant Notice (the “Grant Date”).
| 1.2. | Incorporation of Terms of Plan |
The Option is subject to the terms and conditions set forth in this Agreement and the Plan, which is incorporated herein by reference. In the event of any inconsistency between the Plan and this Agreement, the terms of the Plan will control.
| 2. | PERIOD OF EXERCISABILITY |
| 2.1. | Commencement of Exercisability |
The Option will vest and become exercisable according to the vesting schedule in the Grant Notice (the “Vesting Schedule”) except that any fraction of a Share as to which the Option would be vested or exercisable will be accumulated and will vest and become exercisable only when a whole Share has accumulated. Notwithstanding anything in the Grant Notice, the Plan or this Agreement to the contrary, except as otherwise determined by the Administrator or provided in a binding written agreement between Participant and the Company, the Option will immediately expire and be forfeited as to any portion that is not vested and exercisable as of Participant’s Termination of Service for any reason.
| 2.2. | Duration of Exercisability |
The Vesting Schedule is cumulative. Any portion of the Option which vests and becomes exercisable will remain vested and exercisable until the Option expires. The Option will be forfeited immediately upon its expiration.
| 2.3. | Expiration of Option |
The Option may not be exercised to any extent by anyone after, and will expire on, the first of the following to occur:
| (a) | The final expiration date in the Grant Notice; |
| (b) | Except as the Administrator may otherwise approve, the expiration of three (3) months from the date of Participant’s Termination of Service, unless Participant’s Termination of Service is for Cause or by reason of Participant’s death or Disability; |
| (c) | Except as the Administrator may otherwise approve, the expiration of one (1) year from the date of Participant’s Termination of Service by reason of Participant’s Disability; |
| (d) | Except as the Administrator may otherwise approve, the expiration of eighteen (18) months from the date of Participant’s Termination of Service by reason of Participant’s death; |
| (e) | Except as the Administrator may otherwise approve, Participant’s Termination of Service for Cause; |
| (f) | Immediately upon a Corporate Event if the Administrator has determined that the Option will terminate in connection with a Corporate Event; |
| (g) | The day before the tenth anniversary of the Grant Date. |
Notwithstanding the foregoing, if Participant dies during the period provided in Section 2.3(b) or 2.3(c) above, the term of the Option shall not expire until the earlier of (i) eighteen (18) months after Participant’s death, (ii) upon any termination of the Option in connection with a Corporate Event, (iii) the Final Expiration Date indicated in the Grant Notice, or (iv) the day before the tenth anniversary of the Grant Date. Additionally, the post-termination exercise period of the Option may be extended as provided in the Plan.
| 3. | EXERCISE OF OPTION |
| 3.1. | Person Eligible to Exercise |
During Participant’s lifetime, only Participant may exercise the Option. After Participant’s death, any exercisable portion of the Option may, prior to the time the Option expires, be exercised by Participant’s Designated Beneficiary as provided in the Plan.
| 3.2. | Partial Exercise |
Any exercisable portion of the Option or the entire Option, if then wholly exercisable, may be exercised, in whole or in part, according to the procedures in the Plan at any time prior to the time the Option or portion thereof expires, except that the Option may only be exercised for whole Shares.
| 3.3. | Tax Withholding. |
| (a) | The Company has the right and option, but not the obligation, to treat Participant’s failure to provide timely payment in accordance with the Plan of any tax and/or social security withholding obligations arising in connection with the Option as Participant’s election to satisfy all or any portion of the withholding tax by requesting the Company retain Shares otherwise issuable under the Option. |
| (b) | Participant acknowledges that Participant is ultimately liable and responsible for all taxes owed in connection with the Option, regardless of any action the Company or any Subsidiary takes with respect to any tax and/or social security withholding obligations that arise in connection with the Option. Neither the Company nor any Subsidiary makes any representation or undertaking regarding the treatment of any tax and/or social security withholding in connection with the awarding, vesting or exercise of the Option or the subsequent sale of Shares. The Company and the Subsidiaries do not commit and are under no obligation to structure the Option to reduce or eliminate Participant’s tax and/or social security liability. |
| (c) | By accepting the Option, Participant agrees that Participant will not sell, dispose of, transfer, make any short sale of, grant any option for the purchase of, or enter into any hedging or similar transaction with the same economic effect as a sale with respect to any Shares or other securities of the Company held by Participant, for a period of one hundred eighty (180) days following the effective date of a registration statement of the Company filed under the Securities Act or such longer period as the underwriters or the Company will request to facilitate compliance with FINRA Rule 2241 or any successor or similar rules or regulation (the “Lock-Up Period”); provided, however, that nothing contained in this section will prevent the exercise of a repurchase option, if any, in favor of the Company during the Lock-Up Period. Participant further agrees to execute and deliver such other agreements as may be reasonably requested by the Company or the underwriters that are consistent with the foregoing or that are necessary to give further effect thereto. In order to enforce the foregoing covenant, the Company may impose stop-transfer instructions with respect to Participant’s Shares (or other securities of the Company) until the end of such period. Participant also agrees that any transferee of any Shares (or other securities of the Company) held by Participant will be bound by this Section 3.3(c). The underwriters of the Company’s Shares are intended third party beneficiaries of this Section 3.3(c) and will have the right, power and authority to enforce the provisions hereof as though they were a party hereto. |
4. OTHER PROVISIONS
| 4.1. | Option Not a Service Contract. |
By accepting the Option, Participant acknowledges, understands and agrees that:
| (a) | the Option is not an employment or service contract, and nothing in the Option will be deemed to create in any way whatsoever any obligation on Participant’s part to continue in the employ of the Company or any Group Company, or of the Company or any Group Company to continue Participant’s employment. In addition, nothing in Participant’s Option will obligate the Company or any Group Company, their respective shareholders, boards of directors, officers or employees to continue any relationship that Participant might have as a Director or Consultant for the Company or any Group Company; |
| (b) | the Plan is established voluntarily by the Company, it is discretionary in nature, and may be amended, suspended or terminated by the Company at any time, to the extent permitted under the Plan; |
| (c) | the grant of the Option is voluntary and occasional and does not create any contractual or other right to receive future grants of options (whether on the same or different terms), or benefits in lieu of options, even if options have been granted in the past; |
| (d) | Participant’s options and any Shares acquired under the Plan on exercise of Participant’s options, and the income and value of same, are not part of normal or expected compensation for any purpose, including, without limitation, calculating any severance, resignation, termination, redundancy, dismissal, end-of-service payments, bonuses, long-service awards, holiday pay, pension or retirement or welfare benefits or similar payments; |
| (e) | the future value of the Shares underlying the Option is unknown, indeterminable, and cannot be predicted with certainty; |
| (f) | neither the Company nor any Group Company shall be liable for any foreign exchange rate fluctuation between Participant’s local currency and the United States Dollar (or such other currency in which the Exercise Price may be denominated) that may affect the value of Participant’s options or of any amounts due to Participant pursuant to the exercise of the Option or the subsequent sale of any Shares received; |
| (g) | for the purposes of the Option, Participant’s status as a Service Provider will be considered terminated as of the date Participant is no longer actively providing services to the Company or one of its Group Companies (regardless of the reason for such termination and whether or not later found to be invalid or in breach of employment laws in the jurisdiction where Participant is employed or the terms of Participant’s employment agreement, if any), and unless otherwise expressly provided in this Agreement or determined by the Company, (i) Participant’s right to vest in the Option under the Plan, if any, and (ii) the period (if any) during which Participant may exercise the Option after such termination as a Service Provider will terminate as of such date and in each instance will not be extended by any notice period or any period of “garden leave” or similar period mandated under employment laws in the jurisdiction where Participant is employed or the terms of Participant’s employment agreement, if any; and the Board shall have the exclusive discretion to determine when Participant is no longer actively providing services for purposes of the Option (including whether Participant may still be considered to be providing services while on a leave of absence); and |
| (h) | no claim or entitlement to compensation or damages shall arise from forfeiture of this Option resulting from the termination of Participant’s status as a Service Provider (for any reason whatsoever, whether or not later found to be invalid or in breach of employment laws in the jurisdiction where Participant is employed or the terms of his or her employment or service agreement, if any), and in consideration of the grant of this Option to which Participant is otherwise not entitled, Participant irrevocably agrees never to institute any claim against the Company or any Group Company, waives his or her ability, if any, to bring any such claim, and release the Company and any Group Company from any such claim; if, notwithstanding the foregoing, any such claim is allowed by a court of competent jurisdiction, then, by participating in the Plan, Participant shall be deemed irrevocably to have agreed not to pursue such claim and agree to execute any and all documents necessary to request dismissal or withdrawal of such claim. |
| 4.2. | No Advice Regarding Grant; No Liability for Taxes |
The Company is not providing any tax, legal or financial advice, nor is the Company making any recommendations regarding Participant’s participation in the Plan, or his or her acquisition or sale of the underlying Shares. Participant should consult with his or her own personal tax, legal and financial advisors regarding his or her participation in the Plan before taking any action related to the Plan.
As a condition to accepting the Option, Participant hereby (a) agrees to not make any claim against the Company, Group, or any of its officers, Directors, Employees related to tax or social security liabilities arising from the Option or other Company or Group compensation and (b) acknowledges that Participant was advised to consult with Participant’s own personal tax, legal and financial advisors regarding the tax and social security consequences of the Option and has either done so or knowingly and voluntarily declined to do so. Additionally, if Participant is subject to tax in the United States, Participant acknowledges that the Option is exempt from Section 409A only if the exercise price per share is at least equal to the “fair market value” of a Share on the date of grant as determined by the US Internal Revenue Service and there is no other impermissible deferral of compensation associated with the Option. Additionally, as a condition to accepting the Option, Participant agrees not make any claim against the Company, Group, or any of its Officers, Directors, Employees in the event that the US Internal Revenue Service asserts that such exercise price per share is less than the “fair market value” of a Share on the date of grant as subsequently determined by the US Internal Revenue Service.
| 4.3. | Adjustments |
Participant acknowledges that the Option is subject to adjustment, modification and termination in certain events as provided in this Agreement and the Plan.
| 4.4. | Notices |
Any notice to be given under the terms of this Agreement to the Company must be in writing and addressed to the Company in care of the Company’s Secretary at the Company’s principal office or the Secretary’s then-current email address. Any notice to be given under the terms of this Agreement to Participant must be in writing and addressed to Participant (or, if Participant is then deceased, to the person entitled to exercise the Option) at Participant’s last known mailing address or email address in the Company’s personnel files. By a notice given pursuant to this Section, either party may designate a different address for notices to be given to that party. Any notice will be deemed duly given: (i) if sent by email, when actually received; and (ii) if sent by certified mail (return receipt requested) and deposited with postage prepaid in the applicable national mail, when delivered by a nationally recognized express shipping company.
| 4.5. | Titles |
Titles are provided herein for convenience only and are not to serve as a basis for interpretation or construction of this Agreement.
| 4.6. | Conformity to Applicable Laws |
Participant acknowledges that the Plan, the Grant Notice and this Agreement are intended to conform to the extent necessary with all Applicable Laws and, to the extent Applicable Laws permit, will be deemed amended as necessary to conform to Applicable Laws, and this Option may be unilaterally cancelled by the Company (with the effect that all Participant’s rights hereunder lapse with immediate effect) if the Administrator determines in its reasonable discretion that such conformity is not possible or practicable.
| 4.7. | Successors and Assigns |
The Company may assign any of its rights under this Agreement to single or multiple assignees, and this Agreement will inure to the benefit of the successors and assigns of the Company. Subject to the restrictions on transfer set forth in the Plan, this Agreement will be binding upon and inure to the benefit of the heirs, legatees, legal representatives, successors and assigns of the parties hereto.
| 4.8. | Limitations Applicable to Section 16 Persons |
Notwithstanding any other provision of the Plan or this Agreement, if Participant is subject to Section 16 of the Exchange Act, the Plan, the Grant Notice, this Agreement and the Option will be subject to any additional limitations set forth in any applicable exemptive rule under Section 16 of the Exchange Act (including any amendment to Rule 16b-3) that are requirements for the application of such exemptive rule. To the extent Applicable Laws permit, this Agreement will be deemed amended as necessary to conform to such applicable exemptive rule.
| 4.9. | Entire Agreement |
The Plan, the Grant Notice and this Agreement (including any exhibit hereto) constitute the entire agreement of the parties and supersede in their entirety all prior undertakings and agreements of the Company and Participant with respect to the subject matter hereof, with the exception of other equity awards previously granted to Participant and any written employment agreement, offer letter, severance agreement, written severance plan or policy, or other written agreement between the Company and Participant in each case that specifies the terms that should govern this Option.
| 4.10. | Agreement Severable |
In the event that any provision of the Grant Notice or this Agreement is held illegal or invalid, the provision will be severable from, and the illegality or invalidity of the provision will not be construed to have any effect on, the remaining provisions of the Grant Notice or this Agreement.
| 4.11. | Limitation on Participant’s Rights |
Participation in the Plan confers no rights or interests other than as herein provided. This Agreement creates only a contractual obligation on the part of the Company as to amounts payable and may not be construed as creating a trust. Neither the Plan nor any underlying program, in and of itself, has any assets. Participant will have only the rights of a general unsecured creditor of the Company with respect to amounts credited and benefits payable, if any, with respect to the Option, and rights no greater than the right to receive the Shares as a general unsecured creditor with respect to the Option, as and when exercised pursuant to the terms hereof.
| 4.12. | Counterparts |
The Grant Notice may be executed in one or more counterparts, including by way of any electronic signature, subject to Applicable Laws, each of which will be deemed an original and all of which together will constitute one instrument.
| 4.13. | [ISO |
If the Option is designated as an ISO:
| (a) | Participant acknowledges that to the extent the aggregate fair market value of shares (determined as of the time the option with respect to the shares is granted) with respect to which options intended to qualify as “incentive stock options” under Section 422 of the Code, including the Option, are exercisable for the first time by Participant during any calendar year exceeds $100,000 or if for any other reason such options do not qualify or cease to qualify for treatment as “incentive stock options” under Section 422 of the Code, such options (including the Option) will be treated as non-qualified options. Participant further acknowledges that the rule set forth in the preceding sentence will be applied by taking the Option and other options into account in the order in which they were granted, as determined under Section 422(d) of the Code. |
| (b) | Participant also acknowledges that if the Option is exercised more than three (3) months after Participant’s Termination of Service, other than by reason of death or Disability, the Option will be taxed as a Non-Qualified Option. If the Company provides for the extended exercisability of the Option under certain circumstances for Participant’s benefit, the Option will not necessarily be treated as an ISO if Participant exercise the Option more than three (3) months after the date of Participant’s Termination of Service. |
| (c) | Participant will notify the Company in writing within fifteen (15) days after the date of any disposition or other transfer of any Shares acquired under this Agreement if such disposition or other transfer is made (a) within two (2) years from the Grant Date or (b) within one (1) year after the transfer of such Shares to Participant. Such notice will specify the date of such disposition or other transfer and the amount realized, in cash, other property, assumption of indebtedness or other consideration, by Participant in such disposition or other transfer.]8 |
| 4.14. | Choice of Law |
The Agreement and any dispute or claim arising out of or in connection with it or its subject matter or formation (including non-contractual disputes or claims) shall be governed by and construed in accordance with the law of England and Wales disregarding any jurisdiction’s choice-of-law principles requiring the application of a jurisdiction’s laws other than that of England and Wales and the courts of England and Wales shall have exclusive jurisdiction to hear any dispute.
| 4.15. | Other Documents |
Participant hereby acknowledges receipt of or the right to receive a document providing the information required by Rule 428(b)(1) promulgated under the Securities Act, which includes the prospectus document containing the Plan information specified in Section 10(a) of the Securities Act. In addition, Participant acknowledges receipt of the Company’s Insider Trading and Window Period Policy.
| 4.16. | Corporate Events. |
The Option is subject to the terms of any agreement governing a Corporate Event involving the Company, including, without limitation, a provision for the appointment of a shareholder representative that is authorized to act on Participant’s behalf with respect to any escrow, indemnities and any contingent consideration.
8 Delete if ISOs are not provided.
4.17 Non-Exempt U.S. Employees.
The Option, whether or not vested, if granted to an Employee who is a non-exempt employee for purposes of the U.S. Fair Labor Standards Act of 1938, as amended, will not be first exercisable for any Shares until at least six months following the Grant Date. Notwithstanding the foregoing, in accordance with the provisions of the U.S. Worker Economic Opportunity Act, any vested portion of the Option may be exercised earlier than six months following the Grant Date in the event of (i) the Participant’s death or Disability, (ii) a Corporate Event in which the Option is not assumed, continued or substituted, (iii) a Change in Control, or (iv) the Participant’s retirement (as such term may be defined in the Agreement or another applicable agreement or, in the absence of any such definition, in accordance with the Company’s then current employment policies and guidelines). This Section 4.17 is intended to operate so that any income derived by a non-exempt employee in connection with the exercise or vesting of the Option will be exempt from Participant’s regular rate of pay.
APPENDIX 3
OPTION GRANT NOTICE (INTERNATIONAL)
IMMUNOCORE HOLDINGS PLC
2021 EQUITY INCENTIVE PLAN [:NON-EMPLOYEE SUB-PLAN]9
Capitalized terms not specifically defined in this Option Grant Notice (the “Grant Notice”) have the meanings given to them in the 2021 Equity Incentive Plan [:Non-Employee Sub-Plan]10 (as amended from time to time, the “Plan”) of Immunocore Holdings Plc (the “Company”).
The Company has granted to the participant listed below (“Participant”) the option described in this Grant Notice (the “Option”), subject to the terms and conditions of the Plan and the Option Agreement attached as Exhibit A (including any special terms and conditions for the Participant’s country set forth in the attached appendix (the “Appendix” and together, the “Agreement”)), both of which are incorporated into this Grant Notice by reference.
| Participant: | |
| Grant Date: | |
| Exercise Price per Share: | |
| Shares Subject to the Option: | |
| Final Expiration Date: | The day before the [10th] anniversary of the Grant Date |
| Vesting Commencement Date: | |
| Vesting Schedule11: | [1/4 of the total number of Shares under Option shall vest and become exercisable on the first anniversary of the Vesting Commencement Date, and 1/12th of the remaining number of Shares under Option shall vest and become exercisable on each Quarter Date thereafter, subject to Participant remaining continuously a Service Provider as of each such date]. |
| Type of Option | Non-Qualified Option |
By Participant’s signature below, Participant agrees to be bound by the terms of this Grant Notice, the Plan, the Agreement and any Group Company policy that may be applicable to the Participant and the Option from time to time (the “Policies”) [including but not limited to the [Company’s claw-back policy / share retention policy / remuneration policy]]12. Participant has reviewed the Plan, this Grant Notice, the Agreement and the Policies in their entirety, has had an opportunity to obtain the advice of counsel prior to executing this Grant Notice and fully understands all provisions of the Plan, this Grant Notice, the Agreement and the Policies. Participant hereby agrees to accept as binding, conclusive and final all decisions or interpretations of the Administrator upon any questions arising under the Plan, this Grant Notice or the Agreement.
9 Note to draft: For Consultants and Directors who are not Employees
10 Note to draft: For Consultants and Directors who are not Employees
11 Note to draft: Selection of applicable vesting schedule, or determination that a different vesting schedule shall apply, subject to discretion of Administrator.
12 Note to draft: Delete as applicable
By accepting this Option, Participant consents to receive this Grant Notice, the Agreement, the Plan, the Policies and any other Plan-related documents by electronic delivery and to participate in the Plan through an on-line or electronic system established and maintained by the Company or another third party designated by the Company. Counterparts may be delivered via facsimile, electronic mail (including pdf or any electronic signature complying with the US federal ESIGN Act of 2000, Uniform Electronic Transactions Act or other Applicable Law) or other transmission method and any counterpart so delivered will be deemed to have been duly and validly delivered and be valid and effective for all purposes.
| Immunocore HOLDINGS Plc | PARTICIPANT | ||
| By: | |||
| Name | [Participant Name] | ||
| Title: | |||
Exhibit A
OPTION AGREEMENT
Capitalized terms not specifically defined in this Agreement (the definition of which includes any special terms and conditions for the Participant’s country set forth in the Appendix) have the meanings specified in the Grant Notice or, if not defined in the Grant Notice, in the Plan.
| 1. | GENERAL |
| 1.1. | Grant of Option |
The Company has granted to Participant the Option effective as of the grant date set forth in the Grant Notice (the “Grant Date”).
| 1.2. | Incorporation of Terms of Plan |
The Option is subject to the terms and conditions set forth in this Agreement and the Plan, which is incorporated herein by reference. In the event of any inconsistency between the Plan and this Agreement, the terms of the Plan will control.
| 2. | PERIOD OF EXERCISABILITY |
| 2.1. | Commencement of Exercisability |
The Option will vest and become exercisable according to the vesting schedule in the Grant Notice (the “Vesting Schedule”) except that any fraction of a Share as to which the Option would be vested or exercisable will be accumulated and will vest and become exercisable only when a whole Share has accumulated. Notwithstanding anything in the Grant Notice, the Plan or this Agreement to the contrary, except as otherwise determined by the Administrator or provided in a binding written agreement between Participant and the Company, the Option will immediately expire and be forfeited as to any portion that is not vested and exercisable as of Participant’s Termination of Service for any reason.
| 2.2. | Duration of Exercisability |
The Vesting Schedule is cumulative. Any portion of the Option which vests and becomes exercisable will remain vested and exercisable until the Option expires. The Option will be forfeited immediately upon its expiration.
| 2.3. | Expiration of Option |
The Option may not be exercised to any extent by anyone after, and will expire on, the first of the following to occur:
| (a) | The final expiration date in the Grant Notice; |
| (b) | Except as the Administrator may otherwise approve, the expiration of three (3) months from the date of Participant’s Termination of Service, unless Participant’s Termination of Service is for Cause or by reason of Participant’s death or Disability; |
| (c) | Except as the Administrator may otherwise approve, the expiration of one (1) year from the date of Participant’s Termination of Service by reason of Participant’s Disability; |
| (d) | Except as the Administrator may otherwise approve, the expiration of eighteen (18) months from the date of Participant’s Termination of Service by reason of Participant’s death; |
| (e) | Except as the Administrator may otherwise approve, Participant’s Termination of Service for Cause; |
| (f) | Immediately upon a Corporate Event if the Administrator has determined that the Option will terminate in connection with a Corporate Event; |
| (g) | The day before the tenth anniversary of the Grant Date. |
Notwithstanding the foregoing, if Participant dies during the period provided in Section 2.3(b) or 2.3(c) above, the term of the Option shall not expire until the earlier of (i) eighteen (18) months after Participant’s death, (ii) upon any termination of the Option in connection with a Corporate Event, (iii) the Final Expiration Date indicated in the Grant Notice, or (iv) the day before the tenth anniversary of the Grant Date. Additionally, the post-termination exercise period of the Option may be extended as provided in the Plan.
| 3. | EXERCISE OF OPTION |
| 3.1. | Person Eligible to Exercise |
During Participant’s lifetime, only Participant may exercise the Option. After Participant’s death, any exercisable portion of the Option may, prior to the time the Option expires, be exercised by Participant’s Designated Beneficiary as provided in the Plan.
| 3.2. | Partial Exercise |
Any exercisable portion of the Option or the entire Option, if then wholly exercisable, may be exercised, in whole or in part, according to the procedures in the Plan at any time prior to the time the Option or portion thereof expires, except that the Option may only be exercised for whole Shares.
| 3.3. | Tax Withholding. |
| (a) | The Company has the right and option, but not the obligation, to treat Participant’s failure to provide timely payment in accordance with the Plan of any tax and/or social security withholding obligations arising in connection with the Option as Participant’s election to satisfy all or any portion of the withholding tax by requesting the Company retain Shares otherwise issuable under the Option. |
| (b) | Participant acknowledges that Participant is ultimately liable and responsible for all taxes owed in connection with the Option, regardless of any action the Company or any Subsidiary takes with respect to any tax and/or social security withholding obligations that arise in connection with the Option. Neither the Company nor any Subsidiary makes any representation or undertaking regarding the treatment of any tax and/or social security withholding in connection with the awarding, vesting or exercise of the Option or the subsequent sale of Shares. The Company and the Subsidiaries do not commit and are under no obligation to structure the Option to reduce or eliminate Participant’s tax and/or social security liability. |
| (c) | By accepting the Option, Participant agrees that Participant will not sell, dispose of, transfer, make any short sale of, grant any option for the purchase of, or enter into any hedging or similar transaction with the same economic effect as a sale with respect to any Shares or other securities of the Company held by Participant, for a period of one hundred eighty (180) days following the effective date of a registration statement of the Company filed under the Securities Act or such longer period as the underwriters or the Company will request to facilitate compliance with FINRA Rule 2241 or any successor or similar rules or regulation (the “Lock-Up Period”); provided, however, that nothing contained in this section will prevent the exercise of a repurchase option, if any, in favor of the Company during the Lock-Up Period. Participant further agrees to execute and deliver such other agreements as may be reasonably requested by the Company or the underwriters that are consistent with the foregoing or that are necessary to give further effect thereto. In order to enforce the foregoing covenant, the Company may impose stop-transfer instructions with respect to Participant’s Shares (or other securities of the Company) until the end of such period. Participant also agrees that any transferee of any Shares (or other securities of the Company) held by Participant will be bound by this Section 3.3(c). The underwriters of the Company’s Shares are intended third party beneficiaries of this Section 3.3(c) and will have the right, power and authority to enforce the provisions hereof as though they were a party hereto. |
| 4. | OTHER PROVISIONS |
| 4.1. | Option Not a Service Contract. |
By accepting the Option, Participant acknowledges, understands and agrees that:
| (a) | the Option is not an employment or service contract, and nothing in the Option will be deemed to create in any way whatsoever any obligation on Participant’s part to continue in the employ of the Company or any Group Company, or of the Company or any Group Company to continue Participant’s employment. In addition, nothing in Participant’s Option will obligate the Company or any Group Company, their respective shareholders, boards of directors, officers or employees to continue any relationship that Participant might have as a Director or Consultant for the Company or any Group Company; |
| (b) | the Plan is established voluntarily by the Company, it is discretionary in nature, and may be amended, suspended or terminated by the Company at any time, to the extent permitted under the Plan; |
| (c) | the grant of the Option is voluntary and occasional and does not create any contractual or other right to receive future grants of options (whether on the same or different terms), or benefits in lieu of options, even if options have been granted in the past; |
| (d) | Participant’s options and any Shares acquired under the Plan on exercise of Participant’s options, and the income and value of same, are not part of normal or expected compensation for any purpose, including, without limitation, calculating any severance, resignation, termination, redundancy, dismissal, end-of-service payments, bonuses, long-service awards, holiday pay, pension or retirement or welfare benefits or similar payments; |
| (e) | the future value of the Shares underlying the Option is unknown, indeterminable, and cannot be predicted with certainty; |
| (f) | neither the Company nor any Group Company shall be liable for any foreign exchange rate fluctuation between Participant’s local currency and the United States Dollar (or such other currency in which the Exercise Price may be denominated) that may affect the value of Participant’s options or of any amounts due to Participant pursuant to the exercise of the Option or the subsequent sale of any Shares received; |
| (g) | for the purposes of the Option, Participant’s status as a Service Provider will be considered terminated as of the date Participant is no longer actively providing services to the Company or one of its Group Companies (regardless of the reason for such termination and whether or not later found to be invalid or in breach of employment laws in the jurisdiction where Participant is employed or the terms of Participant’s employment agreement, if any), and unless otherwise expressly provided in this Agreement or determined by the Company, (i) Participant’s right to vest in the Option under the Plan, if any, and (ii) the period (if any) during which Participant may exercise the Option after such termination as a Service Provider will terminate as of such date and in each instance will not be extended by any notice period or any period of “garden leave” or similar period mandated under employment laws in the jurisdiction where Participant is employed or the terms of Participant’s employment agreement, if any; and the Board shall have the exclusive discretion to determine when Participant is no longer actively providing services for purposes of the Option (including whether Participant may still be considered to be providing services while on a leave of absence); and |
| (h) | no claim or entitlement to compensation or damages shall arise from forfeiture of this Option resulting from the termination of Participant’s status as a Service Provider (for any reason whatsoever, whether or not later found to be invalid or in breach of employment laws in the jurisdiction where Participant is employed or the terms of his or her employment or service agreement, if any), and in consideration of the grant of this Option to which Participant is otherwise not entitled, Participant irrevocably agrees never to institute any claim against the Company or any Group Company, waives his or her ability, if any, to bring any such claim, and release the Company and any Group Company from any such claim; if, notwithstanding the foregoing, any such claim is allowed by a court of competent jurisdiction, then, by participating in the Plan, Participant shall be deemed irrevocably to have agreed not to pursue such claim and agree to execute any and all documents necessary to request dismissal or withdrawal of such claim. |
| 4.2. | No Advice Regarding Grant; No Liability for Taxes |
The Company is not providing any tax, legal or financial advice, nor is the Company making any recommendations regarding Participant’s participation in the Plan, or his or her acquisition or sale of the underlying Shares. Participant should consult with his or her own personal tax, legal and financial advisors regarding his or her participation in the Plan before taking any action related to the Plan.
As a condition to accepting the Option, Participant hereby (a) agrees to not make any claim against the Company, Group, or any of its officers, Directors, Employees related to tax or social security liabilities arising from the Option or other Company or Group compensation and (b) acknowledges that Participant was advised to consult with Participant’s own personal tax, legal and financial advisors regarding the tax and social security consequences of the Option and has either done so or knowingly and voluntarily declined to do so. Additionally, if Participant is subject to tax in the United States, Participant acknowledges that the Option is exempt from Section 409A only if the exercise price per share is at least equal to the “fair market value” of a Share on the date of grant as determined by the US Internal Revenue Service and there is no other impermissible deferral of compensation associated with the Option. Additionally, as a condition to accepting the Option, Participant agrees not make any claim against the Company, Group, or any of its Officers, Directors, Employees in the event that the US Internal Revenue Service asserts that such exercise price per share is less than the “fair market value” of a Share on the date of grant as subsequently determined by the US Internal Revenue Service.
| 4.3. | Adjustments |
Participant acknowledges that the Option is subject to adjustment, modification and termination in certain events as provided in this Agreement and the Plan.
| 4.4. | Language |
Participant acknowledges that he or she is sufficiently proficient in the English language, or has consulted with an advisor who is sufficiently proficient in English, so as to allow him or her to understand the terms and conditions of this Agreement. If Participant has received this Agreement, or any other document related to the Option and/or the Plan translated into a language other than English and if the meaning of the translated version is different than the English version, the English version will control.
| 4.5. | Foreign Assets/Account, Exchange Control and Tax Reporting |
Participant may be subject to foreign asset/account, exchange control and/or tax reporting requirements as a result of the acquisition, holding and/or transfer of Shares or cash (including dividends and the proceeds arising from the sale of Shares) derived from Participant’s participation in the Plan in, to and/or from a brokerage/bank account or legal entity located outside Participant’s country. The applicable laws in Participant’s country may require that he or she report such accounts, assets and balances therein, the value thereof and/or the transactions related thereto to the applicable authorities in such country. Participant may also be required to repatriate sale proceeds or other funds received as a result of his or her participation in the Plan to his or her country through a designated bank or broker within a certain time after receipt. Participant acknowledges that it is his or her responsibility to be compliant with such regulations and he or she is encouraged to consult with his or her personal legal advisor for any details.
| 4.6. | Appendix |
Notwithstanding any provisions in this Agreement, the Option shall be subject to the special terms and conditions for Participant’s country set forth in the Appendix attached to this Agreement. Moreover, if Participant relocates to one of the countries included therein, the terms and conditions for such country will apply to Participant to the extent the Company determines that the application of such terms and conditions is necessary or advisable for legal or administrative reasons. The Appendix constitutes part of this Agreement.
| 4.7. | Notices |
Any notice to be given under the terms of this Agreement to the Company must be in writing and addressed to the Company in care of the Company’s Secretary at the Company’s principal office or the Secretary’s then-current email address. Any notice to be given under the terms of this Agreement to Participant must be in writing and addressed to Participant (or, if Participant is then deceased, to the person entitled to exercise the Option) at Participant’s last known mailing address or email address in the Company’s personnel files. By a notice given pursuant to this Section, either party may designate a different address for notices to be given to that party. Any notice will be deemed duly given: (i) if sent by email, when actually received; and (ii) if sent by certified mail (return receipt requested) and deposited with postage prepaid in the applicable national mail, when delivered by a nationally recognized express shipping company.
| 4.8. | Titles |
Titles are provided herein for convenience only and are not to serve as a basis for interpretation or construction of this Agreement.
| 4.9. | Conformity to Applicable Laws |
Participant acknowledges that the Plan, the Grant Notice and this Agreement are intended to conform to the extent necessary with all Applicable Laws and, to the extent Applicable Laws permit, will be deemed amended as necessary to conform to Applicable Laws, and this Option may be unilaterally cancelled by the Company (with the effect that all Participant’s rights hereunder lapse with immediate effect) if the Administrator determines in its reasonable discretion that such conformity is not possible or practicable.
| 4.10. | Successors and Assigns |
The Company may assign any of its rights under this Agreement to single or multiple assignees, and this Agreement will inure to the benefit of the successors and assigns of the Company. Subject to the restrictions on transfer set forth in the Plan, this Agreement will be binding upon and inure to the benefit of the heirs, legatees, legal representatives, successors and assigns of the parties hereto.
| 4.11. | Limitations Applicable to Section 16 Persons |
Notwithstanding any other provision of the Plan or this Agreement, if Participant is subject to Section 16 of the Exchange Act, the Plan, the Grant Notice, this Agreement and the Option will be subject to any additional limitations set forth in any applicable exemptive rule under Section 16 of the Exchange Act (including any amendment to Rule 16b-3) that are requirements for the application of such exemptive rule. To the extent Applicable Laws permit, this Agreement will be deemed amended as necessary to conform to such applicable exemptive rule.
| 4.12. | Entire Agreement |
The Plan, the Grant Notice and this Agreement (including any exhibit hereto) constitute the entire agreement of the parties and supersede in their entirety all prior undertakings and agreements of the Company and Participant with respect to the subject matter hereof, with the exception of other equity awards previously granted to Participant and any written employment agreement, offer letter, severance agreement, written severance plan or policy, or other written agreement between the Company and Participant in each case that specifies the terms that should govern this Option.
| 4.13. | Agreement Severable |
In the event that any provision of the Grant Notice or this Agreement is held illegal or invalid, the provision will be severable from, and the illegality or invalidity of the provision will not be construed to have any effect on, the remaining provisions of the Grant Notice or this Agreement.
| 4.14. | Limitation on Participant’s Rights |
Participation in the Plan confers no rights or interests other than as herein provided. This Agreement creates only a contractual obligation on the part of the Company as to amounts payable and may not be construed as creating a trust. Neither the Plan nor any underlying program, in and of itself, has any assets. Participant will have only the rights of a general unsecured creditor of the Company with respect to amounts credited and benefits payable, if any, with respect to the Option, and rights no greater than the right to receive the Shares as a general unsecured creditor with respect to the Option, as and when exercised pursuant to the terms hereof.
| 4.15. | Counterparts |
The Grant Notice may be executed in one or more counterparts, including by way of any electronic signature, subject to Applicable Laws, each of which will be deemed an original and all of which together will constitute one instrument.
| 4.16. | Choice of Law |
The Agreement and any dispute or claim arising out of or in connection with it or its subject matter or formation (including non-contractual disputes or claims) shall be governed by and construed in accordance with the law of England and Wales disregarding any jurisdiction’s choice-of-law principles requiring the application of a jurisdiction’s laws other than that of England and Wales and the courts of England and Wales shall have exclusive jurisdiction to hear any dispute.
| 4.17. | Other Documents |
Participant hereby acknowledges receipt of or the right to receive a document providing the information required by Rule 428(b)(1) promulgated under the Securities Act, which includes the prospectus document containing the Plan information specified in Section 10(a) of the Securities Act. In addition, Participant acknowledges receipt of the Company’s Insider Trading and Window Period Policy.
| 4.18. | Corporate Events. |
The Option is subject to the terms of any agreement governing a Corporate Event involving the Company, including, without limitation, a provision for the appointment of a shareholder representative that is authorized to act on Participant’s behalf with respect to any escrow, indemnities and any contingent consideration.
APPENDIX TO OPTION AGREEMENT
This Appendix includes special terms and conditions that govern the Option granted to Participant under the Plan if Participant resides and/or works in one of the countries listed below.
The information contained herein is general in nature and may not apply to Participant’s particular situation, and Participant is advised to seek appropriate professional advice as to how the relevant laws in Participant’s country may apply to his or her situation. If Participant is a citizen or resident of a country other than the one in which he or she is currently working and/or residing, transfers employment and/or residency to another country after the Grant Date, is a Consultant, changes employment status to a consultant position, or is considered a resident of another country for local law purposes, the Company shall, in its discretion, determine the extent to which the special terms and conditions contained herein shall be applicable to Participant. References to an employer (if any) shall include any entity that engages Participant’s services.
Italy
Stock Option Exercises. Due to regulatory requirements, notwithstanding any other provision of the Plan or the Agreement, Participant will be required to exercise the Option using a cashless sell-all exercise method, pursuant to which all Shares subject to the exercised Option will be sold immediately upon exercise and the proceeds of sale, less the exercise price, any tax and/or social security withholding obligations and broker’s fees or commissions, will be remitted to Participant in cash in accordance with any applicable exchange control laws and regulations. Participant will not be permitted to hold Shares after exercise. The Company reserves the right to provide additional methods of exercise depending on the development of local laws.
Plan Acknowledgement. Participant acknowledges that he or she has read and specifically and expressly approve the following sections of the Agreement: (3.3) Tax Withholding; (4.1) Option Not a Service Contract; (4.4) Language; (4.13) Agreement Severable; (4.14) Limitation on Participant’s Rights; (4.16) Choice of Law; and (4.17) Other Documents; Section 10(j)(ii) of the Plan; and the Italy country-specific terms and conditions of this Appendix.
Foreign Asset/Account Reporting Information. If Participant is an Italian resident and, during any fiscal year, holds investments or financial assets outside of Italy (e.g., cash, Shares) which may generate income taxable in Italy (or if Participant is the beneficial owner of such an investment or asset even if Participant does not directly hold the investment or asset), Participant is required to report such investments or assets on Participant’s annual tax return for such fiscal year (on UNICO Form, RW Schedule, or on a special form if Participant is not required to file a tax return).
Foreign Financial Assets Tax. The fair market value of any Shares held outside of Italy is subject to a foreign assets tax. Financial assets include Shares acquired under the Plan. The taxable amount will be the fair market value of the financial assets assessed at the end of the calendar year. The Participant should consult with his or her personal tax advisor about the foreign financial assets tax.
Poland
Data Privacy. The privacy notice mentioned in Section 10(j)(ii) of the Plan, regarding processing of information relating to Participant, is available at [13]. The Participant acknowledges that his or her personal data will be processed on the basis Article 6(1)(b) of the EU General Data Protection Regulation for the purpose of performing the Agreement.
Tax. By accepting the Option and accepting the terms of the Agreement, the Participant acknowledges and agrees to comply with all applicable Polish laws and report any income and pay any and all applicable taxes and other mandatory contributions, as required by Polish laws, associated with the Option, the sale of Shares acquired under the Plan, and the receipt of any dividends paid on such Shares. The Company accepts no obligation in this respect.
Exchange Control Information. Polish residents holding foreign securities which are not connected with their business (including Shares) must report information to the National Bank of Poland on transactions and balances of the securities deposited in such accounts if the value of such transactions or balances (calculated individually or together with other assets or liabilities held abroad) exceeds PLN 7,000,000. If required, the reports are due on a quarterly basis. Polish entrepreneurs are also required to transfer funds through a bank account or payment institution in Poland if: (i) the payment is connected with their business, and (ii) the transferred amount in any single transaction exceeds PLN 15,000, and (iii) the other party to the transaction is also an entrepreneur. Further, upon the request of a Polish bank, Polish residents are required to inform the bank about all foreign exchange transactions performed through such bank. In addition, Polish residents are required to store documents connected with any foreign exchange transaction for a period of 5 years from the end of the year in which such transaction was made. Penalties may apply for failure to comply with exchange control requirements.
Securities Law Information. The grant of the Option is not intended to be publicly offered in or from Poland and therefore it is not subject to securities registration in Poland. No document or material related to the Plan has been or will be filed with, approved or supervised by any Polish regulatory authority. No such document or material may be made publicly available in Poland.
Niniejszym potwierdzają Państwo, że znają Państwo język angielski w stopniu umożliwiającym pełne zrozumienie dokumentów dotyczących Planu Opcyjnego, w tym w szczególności Umowy Opcyjnej wraz z załącznikami, lub też skonsultowali Państwo treść tych dokumentów z odpowiednim doradcą. Jeżeli otrzymali Państwo jakikolwiek dokument dotyczący Planu Opcyjnego przetłumaczony na inny język niż angielski, angielska wersja językowa tego dokumentu będzie przeważająca.
13 Link to HR intranet or similar where privacy notices / information can be found to be included.
11
|
Subsidiary
|
|
Jurisdiction
|
||
|
Immunocore Limited
|
|
England and Wales
|
||
|
Immunocore Nominees Limited
|
|
England and Wales
|
||
|
Immunocore Ireland Limited
|
|
Republic of Ireland
|
||
|
Immunocore, LLC
|
|
Delaware
|
||
|
Immunocore Commercial LLC
|
|
Delaware
|
||
Exhibit 12.1
|
1.
|
I have reviewed this annual report on Form 20-F of Immunocore Holdings plc (the “Company”);
|
|
2.
|
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not
misleading with respect to the period covered by this report;
|
|
3.
|
Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the Company as of, and for,
the periods presented in this report;
|
|
4.
|
The Company’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) for
the Company and have:
|
|
(a)
|
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the Company, including its consolidated subsidiaries,
is made known to us by others within those entities, particularly during the period in which this report is being prepared;
|
|
(b)
|
Evaluated the effectiveness of the Company’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this
report based on such evaluation; and
|
|
(c)
|
Disclosed in this report any change in the Company’s internal control over financial reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the
Company’s internal control over financial reporting; and
|
|
5.
|
The Company’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the Company’s auditors and the audit committee
of the Company’s board of directors (or persons performing the equivalent functions):
|
|
(a)
|
All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the Company’s ability to record, process, summarize and report
financial information; and
|
|
(b)
|
Any fraud, whether or not material, that involves management or other employees who have a significant role in the Company’s internal control over financial reporting.
|
|
By:
|
/s/ Bahija Jallal, Ph.D. | |
|
Bahija Jallal, Ph.D.
|
||
|
Chief Executive Officer
|
||
|
(Principal Executive Officer)
|
Exhibit 12.2
|
1.
|
I have reviewed this annual report on Form 20-F of Immunocore Holdings plc (the “Company”);
|
|
2.
|
Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not
misleading with respect to the period covered by this report;
|
|
3.
|
Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the Company as of, and for,
the periods presented in this report;
|
|
4.
|
The Company’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) for the Company and have:
|
|
(a)
|
Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the Company, including its consolidated subsidiaries,
is made known to us by others within those entities, particularly during the period in which this report is being prepared;
|
|
(b)
|
Evaluated the effectiveness of the Company’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this
report based on such evaluation; and
|
|
(c)
|
Disclosed in this report any change in the Company’s internal control over financial reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the
Company’s internal control over financial reporting; and
|
|
5.
|
The Company’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the Company’s auditors and the audit committee of the Company’s board of directors (or
persons performing the equivalent functions):
|
|
(a)
|
All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the Company’s ability to record, process, summarize and report
financial information; and
|
|
(b)
|
Any fraud, whether or not material, that involves management or other employees who have a significant role in the Company’s internal control over financial reporting.
|
|
By:
|
/s/ Brian Di Donato | |
|
Brian Di Donato
|
||
|
Chief Financial Officer
|
||
|
(Principal Financial Officer)
|
Exhibit 13.1
|
(1)
|
The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended; and
|
|
(2)
|
The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
|
|
/s/ Bahija Jallal, Ph.D.
|
||
|
Chief Executive Officer
|
||
|
(Principal Executive Officer)
|
|
/s/ Brian Di Donato
|
||
|
Chief Financial Officer
|
||
|
(Principal Financial Officer)
|